1. Introduction
mTOR represents the target of a molecule, known as sirolimus or rapamycin, produced by
Streptomyces hygroscopicus [
1]. This macrolide represents a highly conserved serine/threonine-protein kinase, which integrates multiple signals from extracellular and intracellular signaling pathways responsible for the regulation of many fundamental cells and biological processes related to metabolism, autophagy, mRNA translation, cell growth, survival, ribosome biogenesis, immune function, obesity, diabetes, and aging [
2]. mTOR is an atypical protein kinase belonging to the phosphoinositide-3-kinase (PI3K) family and is commonly organized into two known signaling complexes, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2) [
3]. The three main mTORC1 elements are mTOR, raptor (mTOR-associated regulatory protein), and mLST8 (mammalian lethal protein 8 with Sec13, also known as GβL). Additionally, mTORC1 consists of the two inhibitory subunits: proline-rich Akt substrate of 40 kDa (PRAS40) and DEP domain-containing mTOR-interacting protein (DEPTOR). In contrast, mTORC2 consists of mLST8, mTOR, rictor (an mTOR-independent raptor companion), and mSIN1 (mammalian stress-activated protein kinase interaction protein 1), protor-1/2 (observed with rictor protein composition-1/2), and DEPTOR [
2]. While these two complexes share the same catalytic-TOR subunit, they phosphorylate distinct downstream targets and exhibit different cellular functions.
With the cooperation of the critical elements of mTORC1 and mTORC2, the mTOR signaling system is able to catalyze the phosphorylation of many targets, namely type-I insulin-like growth factor receptor (IGF-IR), protein kinase C (PKC), protein kinase B (Akt), eukaryotic translation initiation factor 4E binding protein 1 (4E-BP1), and ribosomal protein S6 kinase β-1 (S6K1). Thus, mTOR promotes mitochondria-related protein synthesis, suppresses the catabolic process of autophagy, contributes to lipid metabolism, engages in ribosome formation, and acts as a key regulator of mRNA translation [
4,
5,
6]. Although mTOR signaling plays several evolutionarily conserved roles, it remains one of the signaling systems most involved in the tumor process, such as cell cycle, cell proliferation, and apoptosis. Indeed, mTORC1 promotes cancer genesis and metastasis by promoting autophagy and reversing glycolysis.
The mTOR signaling pathway may be proposed as a cancer biomarker, and its targeting constitutes a major therapeutic challenge. Indeed, the mTOR signaling dysregulation is involved in multiple human cancers, including lung, breast, cervical, colon, reproductive, and bone cancers, as well as nasopharyngeal carcinoma. Although previous work has highlighted the role of mTOR in the process of cancerization, the specific involvement of mTOR signaling in the different types of cancer, as well as the pathways of mTOR inhibition, remain poorly discussed. In this context, to determine new pharmacological approaches, researchers have already initiated the targeting of this pathway by using mTOR inhibitors alone or combined with therapies. In combination with natural compounds, mTOR inhibitors were used as a promising source of effective anticancer drugs such as apigetrin, dihydromyricetin, piperlongumine, thymoquinone, glycyrrhizic acid, cryptotanshinone, cannabisin B, licochalcone A, and curcumin [
7]. These agents affect the modulation of autophagy via PI3K/Akt/mTOR pathway inhibition in different cancer cells [
8]. Synthetic compounds also act as cancer-preventive therapeutics targeting the mTOR signaling pathway.
On the other hand, immunotherapy has attracted much attention to the development of targeted therapies inhibiting the PI3K-AKT-mTOR signaling network, which is dysfunctional in several forms of cancer. Recent studies have shown that mTOR plays a crucial role in immune system modulation. Interestingly, mTOR regulates T cells, tumor-associated macrophages (TAMs), and antigen-presenting cells and promotes their development and activation [
9]. In addition, studies have demonstrated the modulation effects of mTORC1/2 on various human cells such as CD4, CD8, Treg, TAMs, cancer-associated fibroblasts (CAF), endothelial cells, and myeloid-derived suppressor cells (MDSCs) [
10,
11]. Based on these findings, there is increasing evidence that targeting the PI3K-AKT-mTOR signaling pathway may affect host immunity and cancer cells. Clinical advances in cancer immunotherapies that target immune checkpoint receptors (CTLA-4 and PD-1) have shown the relevance of immunoevasion as a specificity of malignancy and cancer [
12,
13].
2. mTOR and Cancer
mTOR, MAPK, and nuclear factor kappa B (NF-κB) are among the signaling pathways involved in cancer formation. The mTOR signaling system, in particular, is involved in apoptosis, cell cycle, and cancer cell proliferation [
63]. Surprisingly, mTORC1 has modulated the Warburg effect, which allows cells to survive in low-oxygen environments. However, most tumors do not have TSC1 or TSC2 genetic abnormalities, and mTORC1 activation is caused by p53 inactivation mutations [
64,
65]. Furthermore, p53 keeps mTORC1 active through transactivation of its negative regulators, AMPK1 and TSC2 [
66,
67]. Downstream, the function of mTOR in carcinogenesis is coupled to various metabolic regulatory network levels activating glycolysis by boosting pyruvate kinase expression [
67]. Cellular responses to existing inhibitors are diverse because of the mTOR signaling network, which includes two functionally distinct mTOR complexes, parallel regulatory pathways, and feedback loops [
68].
mTOR may be phosphorylated after Akt activation, and activation of this downstream pathway may be crucial in controlling cell proliferation and differentiation, protein synthesis, and cell metabolism [
69]. As already mentioned, mTOR comprises two molecular complexes; mTORC1 and mTOR2, each with its function. Indeed, mTORC2 phosphorylates additional downstream targets, which can cause Akt feedback, among other things, resulting in a limited influence on protein synthesis and cell growth. Growth hormones not only activate mTORC1, but it is also engaged in cellular stress, and it is linked to autophagy in particular [
70,
71,
72]. Bourneville mutations cause tuberous sclerosis syndrome in the TSC protein TSC2, which suppresses mTOR action under hypoxic and energy-depleted circumstances [
24,
73].
Furthermore, mutations in the phosphatase and tensin homolog (PTEN) gene are the most well-known genetic abnormalities impacting mTOR signaling observed in human cancer [
74]. PTEN mutations are linked to various malignancies, including endometrial cancer, melanoma, bladder, lung, breast, prostate, thyroid cancer, brain cancer, kidney cancer, and others, making it one of the most mutated tumor suppressor genes [
75,
76,
77]. Up to 80% of human malignancies include either gain-of-function or loss-of-function mutations in either the oncogenic Raf, Ras, Akt or PI3K signaling pathways or in the tumor-suppressor genes NF1, PTEN, or TSC [
78].
2.1. Signalling Upstream of mTOR
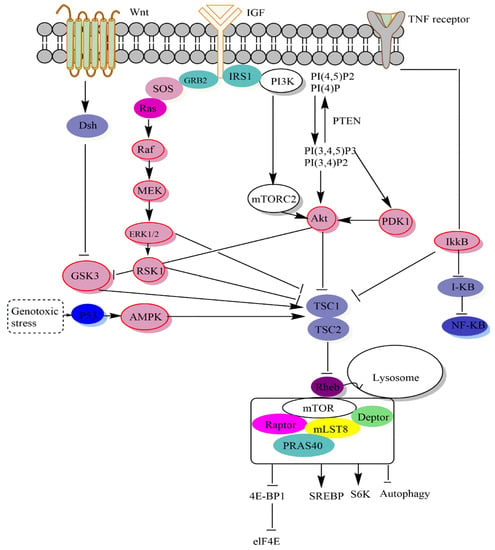
The upstream signals they include, the substrates they control, the biological processes they oversee, and the unique protein compositions of mTOR complexes all contribute to their varying degrees of sensitivity to rapamycin [
3]. Bypass methods outside the PI3K-Akt-mTOR pathway allow growth hormones to affect mTORC1 (
Figure 2). By phosphorylating extracellular kinase (Erk), growth hormones like epidermal growth factor (EGF) may inhibit TSC activity and stimulate mTORC1 [
79]. mTORC1 activity is regulated by many upstream signals that converge on the TSC. Akt [
80], extracellular signal-regulated kinase (ERK) [
79], p90 ribosomal S6 kinase 1 (RSK1) [
27], IκB kinase
β (IKK
β) [
81], and MAPKAPK2 (MK2) [
82] can inhibit and phosphorylate TSC2 [
28] to activate mTORC1. TSC signaling is a critical signaling pathway that controls mTORC1.
Figure 2. The mTOR pathway. mTORC1, also known as the rapamycin-sensitive complex, is made up of four proteins: mTOR, raptor, mLST8, and PRAS40. TSC1/2-Rheb is mTORC1’s primary upstream regulator. mTORC1 regulates protein translation by phosphorylating S6K1 and 4EBP1 via the TSC1/2-Rheb axis, which integrates cellular energy levels, growth hormones, and Wnt signals. Rheb, TSC1/2, AMPK, Rag, and Akt are the key upstream regulators of mTORC1. When mTORC1 is active at the lysosome membrane, it phosphorylates 4E-BP1, S6K, SREBP, and some autophagy components, all of which influence protein synthesis, lipid and lysosome production, energy metabolism, and autophagy. On the other hand, little is known about mTORC2’s upstream regulators.
Low energy levels activate AMPK during glucose deprivation [
83,
84,
85]. In fact, induction of tumor suppressor protein 53 (TP53), during glucose deprivation, inhibited mTORC1 via DNA damage response pathways [
66]. Phosphatidylinositol-3,4-bisphosphate (PI(3,4)P2) and phosphatidylinositol-3,4,5-trisphosphate (PI(3,4,5,)P3) are two physiologically active lipid-producing proteins that are generated when phosphoinositides are phosphorylated by PI3K at the D3 location. By dephosphorylating phosphoinositides, the tumor suppressor phosphatase PTEN blocks PI3K activity at this location. As a result of its binding to the pleckstrin (PH) homology domain of the serine-threonine kinase Akt, PI(3,4,5,)P3 stimulates Akt’s membrane translocation [
86,
87]. TSC2 also converts Rheb to its inactive GDP-related form, which it uses to adversely regulate mTORC1 activity. Growth hormones, which activate mTORC1, reduce TSC1-TSC2 activity, while genotoxic stress, energy shortage, and oxygen starvation increase TSC1-TSC2 activity and reduce mTORC1. Rag GTPases interact with mTORC1, encouraging its translocation from the cytoplasm to the lysosomal membranes, where Rheb is thought to reside, when amino acids are present [
88]. Two of the most important molecules that mTORC1 phosphorylates in order to control protein synthesis are S6K1, also known as p70 S6 kinase, and 4E-BP1, also known as eukaryotic initiation factor binding protein 4E [
89].
By binding and sequencing initiation factor 4E (eIF4E), the unphosphorylated 4E-BP1 protein slows translation initiation [
16,
90]. In fact, by the sterol response element-binding protein (SREBP) that regulates the expression of metabolic genes responsible for cholesterol and fatty acid formation, mTORC1 stimulates de novo lipid synthesis [
91]. Glycogen synthase kinase 3 (GSK3) may also activate TSC, whereas GSK3 can be inhibited by the upstream Wnt protein signal. Therefore, Wnt protein signals may activate mTORC1 in general [
24,
92]. Compared to mTORC1, our understanding of mTORC2 biology is still quite restricted. Growth factors activate mTORC2 by an unknown mechanism. It affects metabolism, cell survival, and cytology by phosphorylating various AGC kinases, including PKCa, SGK1, and Akt and is resistant to acute rapamycin therapy [
93,
94].
2.2. Signaling Downstream of mTOR
The overall consensus is that Akt activates mTOR, which is downstream. According to this concept, abnormal Akt activation upregulates mTOR signaling, making altered cells more responsive to rapamycin treatment [
2]. Furthermore, mTOR controls protein synthesis by phosphorylating and inactivating the mRNA translation repressor, eukaryotic initiation factor binding protein 4E (4E-BP1), and phosphorylating and activating S6 kinase (S6K1) (
Figure 2). These two mTOR downstream effectors, the phosphorylation of which is blocked in vivo by rapamycin, may be phosphorylated in vitro by recombinant mTOR [
89,
95]. Furthermore, the alanine substitution at Asp 2338 in the mTOR catalytic domain is sufficient to inhibit mTOR kinase activity against S6K1 and 4E-BP1 (in vivo and in vitro) [
89,
96]. In most cases, translation is controlled during the process in which a ribosome is attracted to the 5-terminus of an mRNA, which is positioned at a start codon [
97].
Various translation initiation factors facilitate ribosome binding by guiding the ribosome to the 5′-end of an mRNA. The cap structure (m7GpppN, where “m” denotes a methyl group and “N” represents any nucleotide) at the 5′-end of all mRNAs generated by the nucleus is specifically recognized by the eukaryotic translation initiation factor 4E (eIF4E) [
22]. Furthermore, mTOR signaling controls the phosphorylation of several proteins, the most well-known of which are those that regulate mRNA translation (
Figure 2). Several locations on the eukaryotic initiation factor-binding protein 1 4E (4E-BP1) are phosphorylated. On the other hand, it is unlikely to phosphorylate Thr69/70 in 4E-BP1, even though 4E-BP1 and S6K1 connect to the raptor, mTOR’s partner [
98]. Furthermore, mTORC1 regulates cellular processes, including protein synthesis, lipid synthesis, cholesterol synthesis, mitochondrial metabolism, and autophagy. Recent investigations have found that high-dose rapamycin induces apoptosis in human cancer cell lines, which is related to the suppression of 4E-BP1 phosphorylation and the dissociation of the raptor mTORC1 [
99].
On the other hand, S6K activity appears to be increased in several cancer cell lines and tumors. This is often related to elevated mTOR activity caused by loss-of-function mutations in the tumor suppressors LKB1, PTEN, or TSC. It was also discovered that S6K1 levels could be increased. Amplification of chromosomal region 17q23, which encodes the S6K1 gene, is a characteristic of several malignancies, including meningioma [
100,
101]. Furthermore, via activating SREBP-1 and peroxisome proliferation activator (PPAR), mTORC1 stimulates lipid and cholesterol synthesis [
91,
102]. While mTORC1 inhibits autophagy, it is unclear why rapamycin may activate or inhibit this process, given that S6K1 has a beneficial function in autophagy induction [
103]. Overexpression of S6K1 or eIF4E enhances cell size in the absence of rapamycin, and when co-expressed, they collaborate to further increase cell size. Cell growth is reduced, and the effects of eIF4E on cell size are blocked when a phosphorylation site-defective mutant of 4EBP1 is expressed, which constitutively binds to the eIF4E-Cap complex to prevent translation initiation. These findings reveal that mTOR delivers downstream signals to at least two distinct targets, S6K1 and 4EBP1/eIF4E, both of which are involved in translation regulation and govern mammalian cell growth [
104].
Other studies involve mRNA in mTOR regulating expression and activities and therefore in cell transformation. Indeed, overexpression of miR-513b might dramatically reduce proliferation, invasion, migration and increase apoptosis of small-cell lung cancer (NSCLC). Overexpressing HMGB3 counteracted the effects of miR-513b on proliferation, invasion, migration, and apoptosis in non-small cell lung cancer (NSCLC). MiR-513b may control NSCLC cell proliferation, invasion, migration, and apoptosis through HMGB3, according to a study by Wang et al. [
105]. In addition, Sun et al. reported that miR-99a and miR-100 suppressed mTOR in esophageal epidermal carcinoma cell lines, thereby reducing cell proliferation; thus, the miR-99a/100-mTOR signaling pathway is a potential therapeutic target to induce apoptosis to combat esophageal epidermal carcinoma cell lines [
106]. Meanwhile, miR-381 overexpression suppresses RELN expression, which in turn reduces PI3K/AKT/mTOR signaling pathway activation and hence reduces prostate cancer cell growth and enhances apoptosis and autophagy in these cells [
107]. Functionally, miR-216b was linked to breast cancer growth via inhibiting the mTOR signaling pathway by targeting HK2. Upregulation of miR-216b or HK2 silencing decreased cell viability, migration, and invasion while simultaneously inducing autophagy, cell cycle arrest, and apoptosis. These results suggest that miR-216b suppresses HK2 to disable the mTOR signaling pathway, thereby stopping the development of stomach cancer [
108]. Both miR-100 and miR-99a target FKBP51 and IGF1R/mTOR signaling pathways in vitro, suggesting they may provide a novel therapeutic approach for the treatment of acute lymphoblastic leukemia [
109].
Here, we discuss in depth the underlying mechanisms associating mTOR and different types of cancer:
2.3. Breast Cancer
In general, investigations have shown that mTOR signaling activation is associated with enhanced tumor progression, survival, and invasion, as well as frequently reduced survivability of the affected patient. This is particularly valid for breast cancer [
110,
111].
Krieger KL and colleagues conducted a study to explore how the breast cancer 1 (BRCA1)-mTORC2 interaction is functionally affected in breast cancer. The results of this investigation established that the tBRCT domain of BRCA1 interacts independently with the subunits PRR5, RICTOR, and SIN1 of the mTORC2 complex, causing disruption of the mTORC2 complex. This provides direct evidence that the DNA damage response is coordinated with pro-survival mTORC2-Akt signaling. It is also noteworthy that the RICTOR interaction affinity may be marginally greater than that of SIN1 or PRR5 [
112]. Indeed, breast cancer cells not expressing functional BRCA1 have been shown to be more responsive towards mTOR antagonists. Given these insights, it appears likely that mTOR signalling is critical for BRCA1 responsiveness to DNA damage and therefore that BRCA1-negative breast cancer cells are more responsive to pan-mTOR inhibition [
113].
Wazir U et al. sought to examine the relationship between the mRNA expression of mTORC1, raptor, and rictor and the development of human breast cancer. The findings showed that in breast cancer tissues (
p = 0.0018), ductal tumors (
p = 0.0014), in higher grade tumors (grade 2 vs. 3,
p = 0.047) a higher expression of mTOR was identified and was related to a low overall survival (
p = 0.01). Raptor mRNA expression was substantially increased in tumors than in normal tissues. In background breast tissue, tumors were negatively associated with Nottingham prognostic index (NPI1 vs. 2,
p = 0.03) and tumor grade (grade 1 vs. 3,
p = 0.01). Rictor expression was notably higher and related to better overall survival (
p = 0.037) and disease-free survival (
p = 0.048) [
114]. Also, a strong positive relationship was observed between mTOR and hTERT (
p < 0.00001), as confirmed in another study conducted by Dogan F et al. [
115].
These findings are in accordance with the idea that mTORC1 is a major up-regulator of telomerase in breast cancer.
Although there is evidence that Akt and mTOR signaling can potentiate oncogenic signaling, research has consistently reported that the expression of Rictor, which is essential for mTORC2 signaling, is in fact decreased in breast tumors as opposed to healthy breast tissue [
114].
In addition, they are two major controllers of the activity of the mTOR–raptor complex, namely the LKB1/AMPK and PI3K/Akt pathways, both of which affect the tuberous clerosis complex (TSC). Tuberin phosphorylation by Akt blocks the complex’s ability to interact with the activity of Rheb, thereby boosting the activity of mTOR [
116]. These findings are supported by the fact that mutations that activate the PIK3CA gene (encoding for a PI3K subunit) occur frequently in breast cancer, typically involving mutations centered on the kinase and helical domains [
117]. Other prevalent mTOR upstream mutations are found in AKT, with impairment or mutation of AKT and PTEN loss identified in breast cancer [
118].
On the other hand, considering that triple-negative breast cancer (TNBC) is characterized by the lack of progesterone receptor (PR), estrogen receptor (ER), and HER2 overexpression, in this sense, Walsh et al. used an immunohistochemistry assay to measure the levels of mTOR and phospoho (p)-mTOR in 99 non-TNBCs and 89 TNBCs. They found that the nuclear protein p-mTOR was more prevalent in triple-negative cancers than in non-triple-negative cancers (
p < 0.001), thus suggesting that mTOR could represent a novel target for TNBC management [
119].
Concerning mTOR and ER, the drug tamoxifen is often indicated in patients with estrogen receptor-positive breast cancer, but they develop resistance to this drug [
120]. mTOR seems to play a key function in this resistance, because ERα phosphorylates the mTOR pathway at Ser118, making it hyper-reactive to activation and therefore less susceptible to bind to tamoxifen [
121]. Indeed, the PI3K/Akt/mTOR axis can be used in the long term by breast cancer cells to escape their reliance on ER signaling and consequently enhance resistance to tamoxifen [
122].
Regarding mTOR and HER, and given that HER-2 expression is significant in the over-activating of mTOR signaling in breast cancer, and that HER family receptors can promote PI3K-mTOR signalling [
123], resistance to HER-2 therapies in breast cancer, particularly lapatinib (a dual EGFR (HER-1 and HER-2) inhibitor) and trastuzumab (an antibody-based drug), is significantly correlated with mTOR signaling [
124,
125]. It should be noted that mutations in the PI3K pathway leading to activation of mTOR signaling is a factor leading to drug resistance [
122].
2.4. Lung Cancer
Among the most widespread cancers worldwide is lung cancer. It is considered to be one of the leading causes of death, occurring in millions of individuals every year [
126]. With the aim of discovering new molecular targets in the management of non-small cell lung cancer (NSCLC), research has focused mainly on the link between the PI3K/AKT/mTOR signaling pathway and lung cancer development as it affects many aspects related to cell survival, proliferation, differentiation, motility and growth [
127]. The identification of this route as an important treatment target for lung cancer is primarily based on its pivotal implication in the onset and evolution of this type of cancer [
128]. Deregulation of mTOR signaling is prominent in a wide range of tumors, such as lung cancer [
129]. In fact, it has been reported that in NSCLC, PI3K/AKT/mTOR signaling is most commonly disrupted due to multiple molecular impairments in this pathway, such as activation of mutations and/or overactivation of receptor tyrosine kinases (RTKs), activating mutations and/or amplifications of genes involved in PI3K, and inactivating mutations or deletion of the tumor suppressor PTEN [
127,
128,
130,
131].
Furthermore, tumor suppressor liver kinase B1 (LKB1) as a negative regulator of mTOR signaling was specifically identified as being commonly mutated in lung cancer, highlighting a potential role of the mTOR pathway in the development of lung carcinogenesis [
132].
On the other hand, concerning the relationship between mTOR and the expression of eukaryotic initiation factor-4E (eIF-4E) in lung cancer, there is evidence suggesting an involvement of the mTOR pathway in lung carcinogenesis via its binding to eIF-4E, acting as an oncogene in numerous studies [
133]. In bronchial adenocarcinoma, eIF-4E expression was consistently reported to be increased compared to the normal lung [
134]. In addition, the study by Seki et al. [
135] demonstrated that the expression of eIF4E in adenocarcinomas was 3.4 to 7.4 times higher than in normal lungs and significantly correlated with tumor invasiveness and histological stage.
According to an in vivo study conducted by Frankel et al. [
136], the incidence of premalignant adenomatous hyperplastic lesions was markedly enhanced due to the overexpression of IGF-1, considered an activator key to the Akt/mTOR pathway.
Similar to the Kirsten’s rat sarcoma, viral oncogene homolog (KRAS) and the epidermal growth factor receptor (EGFR) are the frequently detected gene mutation in NSCLC. In a study conducted by Conde and colleagues, they found that EGFR mutations have accumulated in a sub-group of lung cancer patients. In addition, the findings showed EGFR gene amplification in the mutant EGFR tumors, as well as a positive association between either alterations in EGFR or KRAS and mTOR signaling activation [
137]. Furthermore, it was shown that in NSCLC metastases, EphA2 expression was found to be significantly higher. Indeed, the EphA2 mutation appears in lung squamous cell carcinoma and promotes invasion and survival of tumors by activation of the mTOR pathway [
138].
Using immunohistochemistry and immunoblotting, the mTOR pathway was implicated in the pathobiological profiles of 150 lung carcinoma specimens and this was correlated with the downstream and upstream proteins p70S6-kinase (S6K) and Akt, respectively. In fact, mTOR and p-mTOR expressions were reported in 68.7% and 53.3% of tumors, respectively. mTor showed the strongest occurrence in lung adenocarcinomas (89.7%) [
139].
2.5. Colon Cancer
The third most common malignancy in both men and women is colorectal cancer. It has an increased incidence of mortality with a mediocre prognosis [
140]. Previous investigations have reported that hyperactivation of mTOR signaling is one of the major factors contributing to the development of colon cancer [
141,
142,
143].
In this sense, Zhang et al. performed a study to examine the pattern of distribution of components of mTOR signaling in adenomas and colorectal cancer (CRC). The results showed that in colorectal adenomas and high-grade intraepithelial neoplasia CRC glandular elements, components of mTOR signaling, including mTOR, p70s6 K and 4EBP1, were strongly activated. Likewise, in human colorectal adenomas and cancers, ex vivo immunohistochemical studies have demonstrated that mTORC1 signaling functions as a premature signaling step in the tumorigenesis process and contributes to the evolution of healthy cells into a neoplastic state [
144].
In addition to mTORC1, mTORC2 in colon cancer has also been shown to be highly overexpressed and plays an influential role in the pathogenesis of this type of cancer.
Roulin et al. found that in HT29 and LS174T colon cancer cells, rictor downregulation significantly reduces cell proliferation. as shown by cell cycle analysis, inactivation of rictor also resulted in cycle arrest at the G
1 phase. They also noted that LS174T cells deficient in rictor were unable to generate tumors in vivo in a xenograft model [
145]. Moreover, the results published by Gulhati et al. [
146] confirm that high mTORC1 and mTORC2 activity contribute to regulating epithelial–mesenchymal transition (EMT), motility and metastasis of CRCs via RhoA and Rac1 signals. Targeting mTOR inhibited colon cancer growth through the 4EBP1/eIF4E/PUMA pathway [
140].
Using Apc+/Delta716 Cdx2+/− compound mutant mice (a murine model of familial adenomatous polyposis), It was found in research by Aoki et al. [
147] that mTOR pathway activation leads to a deregulation of translation and an enhancement of the G1-S phase, coupled with reduced p27 levels and cyclin E-Cdk2 activation. It was also observed from the results that mTORC1 promotes chromosomal instability (CIN) mediated by anaphase bridging, thereby promoting tumor initiation and progression. The anaphase bridge index (ABI) in colon cancer cells was also increased due to forced activation of mTOR via the upstream regulator Akt.
In a recent study, as shown by bioinformatic analysis, the authors have found that in colorectal tumors, the expression of E3 ubiquitin ligase protein RING finger 167 (RNF167) is decreased and that of STAMBPL1 is increased, acting synergistically to control the level of polyubiquitination of Sestrin2 in relation to leucine accessibility. Sestrin2 ubiquitination facilitates its interaction with GATOR2 and suppresses mTORC1 signaling [
148].
On the other hand, there is evidence that fasting has significant antitumor benefits in different cancers, notably CRC. It has been suggested that an mTOR inhibitor may act synergistically with fasting to inhibit CRC proliferation via Farnesyl-Diphosphate Farnesyltransferase 1 (FDFT1)-mediated AKT/mTOR/HIF1α pathway inhibition [
149].
Atractylenolide I (ATL-1) has been shown to be an effective drug in suppressing colorectal tumor progression, primarily by preventing colorectal cancer cell proliferation through glucose metabolism, apoptosis alteration, and stump-like behavior via the AKT/mTOR signaling pathway [
150]. Furthermore, dual suppression by NVP-BEZ235 of both PI3K and mTORC1/2 signaling has been shown to lead to tumor regression in a genetically engineered mouse model for sporadic CRC [
151]. In addition, there is evidence indicating that the association of PI3K/mTOR and EGFR inhibitors has the potential to enhance treatment performance in patients with KRAS-mutant CRC [
152].
2.6. Head and Neck Cancer
Currently, the sixth most commonly diagnosed cancer worldwide is head and neck squamous cell carcinoma (HNSCC) [
153]. Among many cancers, the development of head and neck cancer is the consequence of genetic changes that deregulate mTOR signaling causing a metabolic rearrangement [
154]. In addition, another type of cancer, HNSCC, is characterized by hyperactivation of the mTOR pathway. Indeed, mTORC1 and mTORC2 represent contributing factors to HNSCC tumorigenesis. In HNSCC, deregulation of mTOR was identified as the most prominent genomic alteration (~80–90% of HNSCC) responsible for abnormal mitogenic signaling, compared to other established pathways such as MAPK and JAK/STAT, hosting mutations less than 10% damage [
154,
155].
In an HNSCCS mouse model, Sun et al. reported that conditional Tgfbr1 and Pten deletion was linked to sporadic tongue tumor formation mediated by mTOR activation [
156]. Additionally, the combined targeting of mTOR and PD-L1 has been shown to enhance tumor growth suppression in a syngeneic mouse model of oral cancer [
157]. Using orthotopic CAL33 xenografts carrying PIK3CA mutations, Bozec et al. investigated the effect of temsirolimus in combination with cetuximab and standard chemotherapeutic drugs such as cisplatin and 5-fluorouracil. The results showed a synergistic action of this combination leading to a nearly total arrest of tumor growth due to the profound involvement of mTOR and EGFR activity [
158].
Currently, there are a few validated mutant genes responsible for the activation of mTOR signaling in HNSCC, one of the pathways affected by these mutations is the PI3K-AKT-mTOR pathway, which is activated by EGFR hyperactivation via some types of genetic and epigenetic strategies. As reported, mTORC1 plays a role in the activation of NF-κB downstream of EGFR/PI3K/Akt signaling. Mechanistically, to accelerate NF-κB signaling, mTORC1 stimulates the activity of nuclear factor kappa-B kinase (IKK) inhibitor. This was justified by the study of Li et al. in which the proliferation of head and neck squamous cell carcinoma is regulated by a positive feedback system implicating EGFR/Akt/mTORC1 and IKK/NF-κB [
159].
On the other hand, the PIK3CA mutation is the frequently identified mutation in HNSCC that activates the PI3K pathway. Indeed, the study by Suda et al. [
160] identified three mutations (2.6%) in PIK3CA and an amplification of the copy number was discovered in 37 cases (32.2%) for PIK3CA. The authors suggested that PIK3CA gene copy number amplification is related to a poorer prognosis in patients with HNSCC without lymph node metastasis. Keysar and colleagues analyzed HNSCC patient-derived xenografts for cancer stem cells (CSCs) and determined the molecular characteristics of tumors induced by smoking and human papillomavirus (HPV) profile (HPV-positive and HPV-negative HNSCC). This research elucidated the impact of deregulated PI3K signaling, such as enhanced SOX2 translation and ALDH expression, leading to increased spheroid and tumor formation. This investigation also noted a downregulation of SOX2 levels following AKT1 (downstream of PI3K) or EIF4E (downstream of mTORC1) silencing, indicating a potential link between SOX2 and PI3K regulation. In addition, SOX2 deletion abolished ALDH transcripts and protein level [
161]. Moreover, it has been reported that the inactivation of PTEN induces hyperactivation of PI3K-mediated mTOR signaling [
162]. PTEN gene mutations are also frequent in HNSCC, which may provide useful predictive markers in patients with tongue cancer [
163]. Further, transforming growth factor-beta-receptor 1 (TGFBR1) may also participate in the deregulation of PI3K-mTOR signaling. Indeed, using the Cre-LoxP system, Bian et al. [
164] elucidated the association of TGF-β signaling and the PI3K-mTOR pathway through conditional suppression of TGFBR1 and PTEN in HNSCC mouse models. The results showed that apoptosis decreased and cell proliferation increased, which in turn increased HNSCC tumor growth.
Moreover, mTOR signaling is reported to be aberrantly activated by HRAS mutants in HNSCC tumors [
165]. In this context, the results of the study by Ruicci et al. [
166] showed that mutant HRAS cells are highly resistant to PI3K suppression and suggested the involvement of MAPK and PI3K signaling pathways intersecting at ERK-TSC2, resulting in persistent mTOR action.
2.7. Cervical Cancer
HPV infection of the cervix has been shown to be involved in the potential for cervical cancer pathogenesis [
167]. The hypothesis that mTOR gene overactivation has a significant impact on the development of human cervical carcinoma was confirmed in a study conducted by Ji et al. Using immunohistochemical analysis, the results showed significantly higher mTOR activity in cervical cancers compared to normal cervical tissue. The expression of mTOR in invasive squamous cell carcinomas of the cervix increases progressively according to the grade of malignancy. Upon activation, the downstream target p70S6K is phosphorylated by mTOR [
168]. It also revealed that in malignant tissues, the expression of p70S6K was markedly increased compared to the normal cervix, and in human cervical squamous cell carcinomas it correlated to a pathological degree. Furthermore, the expression of mTOR and P70S6K was found to be strongly correlated positively [
169]. The study by Faried et al. [
170] aimed to investigate the prognostic and predictive function of molecular overexpression in cisplatin-based neoadjuvant chemotherapy-treated cervical cancer. The outcomes indicate that activated Akt and mTOR proved to be important predictive indicators (
p < 0.05). The investigators also explored activated AKT (p-AKT) and mTOR (p-mTOR) expression among patients diagnosed with cervical adenocarcinoma. The findings demonstrated in 50% and 53% of cervical adenocarcinomas, respectively, that p-AKT and p-mTOR were determined. Overall, p-mTOR expression was reported to be an independent and predictive indicator of cervical adenocarcinoma.
In another study by Feng et al., morphoproteomic assays indicate constitutive activation and predominant overexpression of the mTOR pathway in both squamous intraepithelial lesions of high grade and cervical squamous cell carcinomas, as demonstrated by enhanced translocation of nuclear pmTOR and p-p70S6K, which are phosphorylated at putative activation sites, Ser2448 and Thr389, respectively. the resulting induced overexpression of the important upstream signal transducer, EGFR; and the enhanced cell cycle related correlates, Skp2 and mitotic indices [
167]. In contrast, the circTPCN/miR-634/mTOR regulatory pathway has been shown to be involved in cervical cancer tumorigenesis [
171]. A study carried out by Liu et al. [
172] showed an ability of CD155 to interfere within a complex formed by AKT, activating the AKT/mTOR/NF-κB pathway and preventing the activation of apoptosis and autophagy in cervical cancer.
2.8. Reproductive Cancer
Ovarian cancer is the most prevalent malignancy of the female reproductive system. Deng et al. [
173] conducted a study to determine the underlying mechanisms required in the chemoresistance of epithelial ovarian cancer (EOC). The findings demonstrated that the expression of markers of epithelial–mesenchymal transition (EMT) and cancer stem cells (CSCs) was dramatically elevated in chemoresistant EOC cells, coupled with the activation of PI3K/Akt/mTOR signaling. Similarly, overexpression of the miR-126-5p increased the growth, migration and invasion of ovarian cancer cells and suppressed their apoptosis. As evidenced by the luciferase assay, miR-126-5p was able to bind directly to PTEN. By targeting PTEN, most likely miR-126-5p could promote the PI3K/Akt/mTOR pathway [
174]. Furthermore, the increase in miR-18b-5p increased the suppressive properties of exosomes on polycystic ovary syndrome progression. There is evidence that miR-18b-5p targets PTEN and may promote the PI3K/Akt/mTOR pathway [
175].
Regarding prostate cancer, the results of Chang et al. reported that radioresistance in prostate cancer is related to epithelial–mesenchymal transition and improved cancer stem cell phenotypes via activation of the PI3K/Akt/mTOR signaling pathway [
176]. Androgen receptor (AR), MAPK, and WNT signaling cascades are among the major interplaying oncogenic signaling cascades that may activate the PI3K-AKT-mTOR pathway and thus promote prostate cancer growth and drug resistance [
177]. The deregulation of the PI3K-AKT-mTOR pathway due to the loss of PTEN is linked to androgen insensitivity and prostate cancer progression. The PI3K and AR signaling pathways are directly linked to the development of prostate cancer [
178].
Furthermore, immunohistochemical analysis of prostate cancer adenocarcinoma revealed that more than 90% of the cases detected the phosphorylated form of AKT of samples and correlated with high Gleason grade of prostate cancer, confirming that aberrant activation of the PI3K/AKT/mTOR pathway is associated with the progression of prostate cancer [
179].
2.9. Bone Cancer
Disturbances in mTOR signaling have been related to the development of malignant diseases, including bone cancer [
180]. It has been published that protein expression levels of p-PI3K/p-Akt/p-mTOR were increased in the periaqueductal gray of rats with bone cancer [
181]. In addition, neurofibromatosis type 1 (NF1) disease is recognized by deficiencies in bone structure combined with enhanced osteoclastogenesis. Ma et al. [
182] in their investigation found that mTOR hyperactivation is involved in dysfunctional major osteoclastogenesis through hyper-proliferation using primary osteoclast-like cells (OCL) derived from a NF1 mouse model (Nf1 heterozygous mice; Nf1+/−).
On the other hand, another pathway by which activation of the mTOR pathway enhances OS cell metastasis is angiogenesis. In fact, activation of the mTOR pathway in these cells promotes cell growth and proliferation, induces cell metastasis, inhibits the intracellular processes of apoptosis, and suppresses autophagy [
183]. Further research on understanding the mechanisms involved in the occurrence of bone cancer through the mTOR pathway is needed.
2.10. Cancers Caused by Different Type of Viruses
There is now conclusive evidence that 10–15% of all malignant tumors are caused by viruses [
184,
185]. In order to immortalize infected cells, HPV replicates by using the host cell’s replication machinery to express the E6/E7/E5 oncoproteins. This immortalization is carried out not only through the inhibition of p53 and Rb tumor suppressors and the reduction of apoptosis, but also, and above all, by the activation of the PI3K/Akt/mTOR pathway [
186]. Researchers in the United States have demonstrated that activation of the Akt /mTOR enzyme may be identified within minutes of exposure of human keratinocytes to HPV-16 pseudovirions [
170,
187].
A critical metabolic sensor in the growth factor receptor (GFR) pathway, which incorporates growth factor signals into cells, is mTOR. Increased nuclear translocation of p-mTOR (Ser2448) and p70S6K (Thr389) is consistent with EGFR upstream signal transducer overexpression, enhanced cycle times and improved mitotic indices [
167]. Two substrates of the mTOR complex 1, 4E-BP1 and p70S6K, are phosphorylated in response to PI3K/Akt/mTOR activation (Thr389). During the early phases of virus–host cell contact, S6K is activated, which induces the translation machinery and suppresses autophagy [
188].
Shrivastava et al. [
189] showed that HCV infection enhances phospho-mTOR and its downstream target 4EBP1, showing that mTOR is not a negative regulator of HCV-induced autophagy. In autophagy-deficient cells, HCV lowers phospho-mTOR, mTOR, and phospho-4EBP1. HCV upregulates Beclin1 and stimulates the mTOR signaling pathway, which may increase hepatocyte development. Conversely, Aravinth et al.[
190] demonstrate that Epstein-Barr virus latent membrane protein 1 (LMP1) increases CD137 expression in Hodgkin Reed–Sternberg cell lines. CD137 expression is induced by LMP1 via the PI3K-AKT-mTOR pathway. These findings provide further evidence for the involvement of Epstein–Barr virus in the development of Hodgkin’s lymphoma.
2.11. Nasopharyngeal Carcinoma
Using immunohistochemistry, Wang et al. [
191] investigated whether there was a correlation between the expression of the proteins p-Akt, p-p70S6K, and p-4EBP1, and the clinicopathologic features of nasopharyngeal cancer (NPC). The findings indicated that the percentage of positive protein expression for p-Akt, p-p70S6K, and p-4EBP1 in NPC was much higher than in noncancerous nasopharyngeal tissue used as control.
Increased expression of AKT, mTOR and P70S6K proteins in NPC tissues were all related to T-stage, N-stage, clinical stage, distant metastasis and differentiation compared to healthy nasopharyngeal mucosal tissues. These findings suggest a link between the AKT/mTOR signaling pathway and the development of NPCs [
192].
2.12. DLBCL
Primary CNS lymphoma (PCNSL) is a form of aggressive non-Hodgkin’s lymphoma that occurs exclusively in the brain and spinal cord (CNS). Diffuse large B-cell lymphomas represent the vast majority of primary CNS lymphomas (DLBCLs). The positive expression levels of p-AKT, p-mTOR, p-S6, and p-4E-BP1 were substantially greater in PCNSL than in the control group. p-mTOR expression is linked to p-AKT, p-S6, and p-4E-BP1. In 18.9% of PCNSL samples, PTEN gene deletion is linked to p-AKT expression [
193].
2.13. Different Type of Lymphomas
The capacity to target the PI3K/AKT pathway with small molecule inhibitors in Burkitt’s lymphoma cell lines was investigated by Bhatti et al. [
194] who also examined the activation of this pathway in a resistant BL cell line model. PI3K/AKT activation was found to be elevated in rituximab- and chemotherapy-resistant cell lines, and inhibition of AKT or PI3K resulted in anti-lymphoma activity in vitro.
In B-cell malignancy, the B-cell receptor signaling pathway is active and the phosphoinositide 3-kinase (PI3K) pathway is the primary mediator of this activation. Additionaly, emerging PI3K blockers like idelalisib and copanlisib have demonstrated remarkable effectiveness in various types of indolent lymphomas, particularly in marginal zone lymphoma [
195].
Patients with follicular lymphoma have been shown to have an unusually high concentration of somatic mutations in RRAGC, as revealed by targeted sequencing. More than 50% of mutations favored coexistence with mutations within ATP6V1B2 and ATP6AP1, both of which are part of vacuolar H-adenosine triphosphate ATPase (v-ATPase) and are reported to be needed for the activation of mTORC1 mediated by amino acids. RagC mutations improved raptor binding and made mTORC1 signaling more robust to amino acid deprivation [
196].
2.14. Non-Mantle Hodgkin’s Cell Lymphoma (MCL)
Yu et al. [
197] showed that nutlin 3A-mediated stabilization and activation of wt-p53 results in G1-S cell cycle arrest and p53-dependent death in MCL cells. They revealed that activation of p53 can significantly downregulate the AKT/mTOR route through an AMPK-dependent mechanism.
This entry is adapted from the peer-reviewed paper 10.3390/cancers14225520