Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Precious Mathebela | -- | 3731 | 2022-11-16 13:49:39 | | | |
| 2 | Amina Yu | + 11 word(s) | 3742 | 2022-11-17 02:23:54 | | | | |
| 3 | Amina Yu | Meta information modification | 3742 | 2022-11-21 07:57:16 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Mathebela, P.; Damane, B.P.; Mulaudzi, T.V.; Mkhize-Khwitshana, Z.L.; Gaudji, G.R.; Dlamini, Z. Microbiome Metagenomics and Epigenomics on Gastric Cancer. Encyclopedia. Available online: https://encyclopedia.pub/entry/34931 (accessed on 24 July 2026).
Mathebela P, Damane BP, Mulaudzi TV, Mkhize-Khwitshana ZL, Gaudji GR, Dlamini Z. Microbiome Metagenomics and Epigenomics on Gastric Cancer. Encyclopedia. Available at: https://encyclopedia.pub/entry/34931. Accessed July 24, 2026.
Mathebela, Precious, Botle Precious Damane, Thanyani Victor Mulaudzi, Zilungile Lynette Mkhize-Khwitshana, Guy Roger Gaudji, Zodwa Dlamini. "Microbiome Metagenomics and Epigenomics on Gastric Cancer" Encyclopedia, https://encyclopedia.pub/entry/34931 (accessed July 24, 2026).
Mathebela, P., Damane, B.P., Mulaudzi, T.V., Mkhize-Khwitshana, Z.L., Gaudji, G.R., & Dlamini, Z. (2022, November 16). Microbiome Metagenomics and Epigenomics on Gastric Cancer. In Encyclopedia. https://encyclopedia.pub/entry/34931
Mathebela, Precious, et al. "Microbiome Metagenomics and Epigenomics on Gastric Cancer." Encyclopedia. Web. 16 November, 2022.
Copy Citation
The gut microbiome plays a pivotal role in the development and progression of gastric cancer. Similar microbes implicated in gastric cancer carcinogenesis have been detected in some of the risk factors of the disease, with microbial dysbiosis as a common root of concern as it promotes carcinogenesis through dysregulation of cancer immunosurveillance and induction of therapeutic resistance. The microbiome plays an important role in gastric cancer (GC) pathological phenotypes and should be taken into consideration when designing personalized cancer therapies.
gastric cancer (GC)
metabolites
microbiome
H. pylori
dysbiosis
epigenomics
personalized therapy
inflammation
1. The Link between Gut Microbiome and Gastric Cancer Risk Factors
One of the proposed cancer prevention strategies is risk factor (RF) reduction. Treatment of the underlying risk factor (RF) can therefore reduce the risk of developing cancer or aid in the treatment of cancer resulting from RF predisposition. A systemic review by Yusefi et al. reported a total of 52 gastric cancer (GC) RFs which were identified and classified according to 9 categories influenced by familial genetics, lifestyle, environment, medication, and exposure to toxins [1]. These categories can be further grouped into two sub-categories; genetic and modifiable, with genetic factors being hereditary while modifiable ones are acquired through lifestyle and can be changed. The most common RFs for GC include Helicobacter pylori (H. pylori) infection, metabolic syndrome, an increased salt intake with a diet low in fiber, as well as male gender (two-fold increase in males than females) [2][3][4][5][6]. Although GC is more common in males, some subtypes such as the MSI and CIMP-H tumors are more prevalent in females [7][8][9]. It was initially thought that GC affects people of an older age (50 to 70 years), however recent findings show an increased incidence in younger individuals [10][11]. The modifiable RFs often lead to epigenetic alterations, and examples of these include toxins, diet, obesity, infection and so on [12]. Figure 1 shows how various RFs can lead to GC.
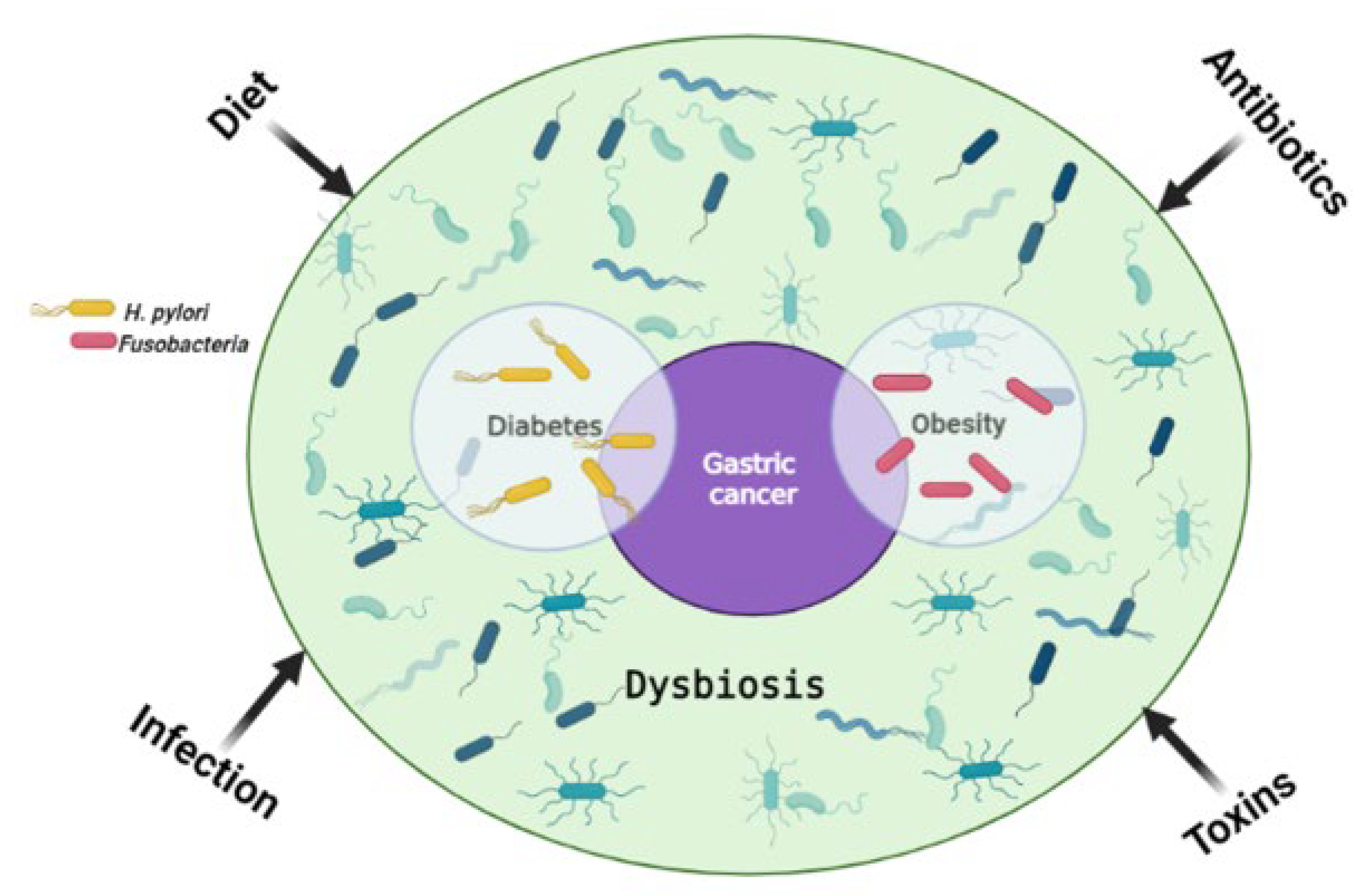
Figure 1. The main risk factors for gastric cancer. Environmental factors influence the gut microbiome and can lead to dysbiosis, one of the main causes of tumorigenesis. H. pylori infection is a shared risk factor between gastric cancer and diabetes, with diabetes being a risk factor for gastric cancer on its own. Similarly, Fusobacteria are a common risk factor for obesity and gastric cancer, with obesity on its own being a risk factor for gastric cancer. Created with BioRender.com. (accessed on 20 September 2022).
1.1. Obesity
The International Agency for Research on Cancer (IARC) regards obesity as the second leading cause of cancer after smoking [13]. About 3–9% of all cancers are approximated to stem from obesity, and GI cancer with obesity origin has the worst prognosis [14][15]. One of the risk factors for obesity includes a high fat and sodium diet, which alters the gut microbiota composition, resulting in gut microbial dysbiosis [16]. Dysbiosis regulates the susceptibility and initiation of many gut malignancies [17]. Kim et al. found that Fusobacterium was enriched in fecal samples of metabolically unhealthy overweight and obese individuals [18]. This shows that the bacteria are a common RF in obesity and GC.
1.2. Diabetes
Diabetes is considered an important contributing factor in GC development, and it is postulated that this is due to shared RFs. These include obesity, a higher infection/reinfection rate, and a lower eradication rate of H. pylori, as well as the chronic use of medication [19]. Additionally, increased salt intake may cooperate with H. pylori infection in the induction of GC and progression. However, a 2022 metanalysis showed no association between diabetes and GC risk in the grading of H. pylori infection and other shared RFs [20]. The authors concluded that diabetes may be associated with excess cardia GC risk.
1.3. Acid Reflux-Related Disorders
Several studies have indicated the association between gastroesophageal reflux disease (GERD) and GC [21][22][23][24]. The overall 5-year survival rate of gastric cardia adenocarcinoma is reported at approximately 31% [25]. Two subtypes of gastric cardia cancer exist; one with GERD origin and the other associated with atrophic gastritis [26]. Misumi et al. defined gastric cardia carcinoma as “a lesion with its center located within 1 cm proximal and 2 cm distal to the esophagogastric mucosal junction” [27]. In a study by Ye et al, it was reported that the risk of developing gastric cardia adenocarcinoma persisted following anti-reflux surgery [24]. This shows that GERD can lead to long-term effects on the stomach mucosa. Some of the risk factors of GERD are obesity and a diet low in fibre, which can have an effect on the gut microbiome [28]. Generally, acid reflux is linked to gut microbiome dysbiosis [29]. A retrospective study by Polat and Polat reported that 82.5% of 1437 GERD patients had H. pylori infection with 1–3 severity score [30], bearing in mind that H. pylori infection is a common RF for GC and GERD.
1.4. Chronic Infection and Inflammation
Infection with pathogenic microbiota leads to the upregulation of inflammatory markers such as cytokines and other secretory proteins. Cytokines such as tumor necrosis factor (TNF), interleukin- 1 (IL-1) and IL-6 expressed within the TME induce cell invasion, metastasis, angiogenesis, growth, and anti-apoptotic effects [31][32][33]. Colonization of H. pylori in the stomach leads to chronic inflammation via the activation of Wnt/β-catenin and other pathways that get activated by the bacteria’s virulence, which further permits the bacteria to survive and thrive in the gut [34][35]. The Wnt/β-catenin signaling pathway is crucial in modulating key cellular processes contributing to carcinogenesis, such as apoptosis, metastasis, proliferation, and genetic stability [36]. Moreover, Wnt/β-catenin has been implicated in pancreatic cancer chemoresistance [37].
The H. pylori commonly infects the stomach, leading to chronic diseases such as peptic ulcer, gastritis, and gastrointestinal (GI) cancers such as GC. The stomach’s naturally acidic environment assists in preventing infection by pathogens. The H. pylori bacteria can maneuver this acidic environment and alter the overall profile of the gastric microbiome [38][39]. There are three mechanisms that the bacteria utilize to alter the GI microbial profile to favor their survival [40]. This includes the employment of enzymes such as ureases which help the bacteria to buffer the acidic pH of the stomach [41]. Secondly, the H. pylori infection effects changes on the cell cycle of gastric epithelial cells, resulting in the elevated expression of p21 and p53 proteins and leading to gene mutations [42][43]. In addition, the infection can lead to abnormal molecular signaling pathways [44]. According to Rossi et al., genomics and proteomics cannot be used to monitor response to therapy [45]. However, a study by Goodman et al. provided evidence that cell-free DNA (cfDNA) can be used to monitor response to chimeric antigen receptor T-cell (CAR-T) therapy in patients with a certain type of B-cell lymphoma [46]. Similarly, the Lewis protein CA-19 is routinely used as a gold standard marker for monitoring response to pancreatic cancer therapies [47][48]. The potential of these “omics” in the area of therapeutics is limited and not well understood.
Gram-negative bacteria including H. pylori are highly resistant to numerous drugs and antibiotics due to the protection provided by their outer membrane [49]. The chronic inflammatory response induced by H. pylori predisposes the mucosal cells to carcinogenesis. In a prospective, double-blind, placebo-controlled, randomized trial published in 2018 by Choi et al., it was observed that GC patients who had either endoscopic resection of early GC or high-grade adenoma, after receiving H. pylori ablation therapy, had lower metachronous GC rates compared to their counterparts who received a placebo [50]. Later on, the same team conducted a randomized trial that was published in 2020 where they evaluated the treatment of H. pylori in first-degree relatives of GC patients [51]. Their results showed that the treatment lowered the risk of developing GC by 55% when compared to the placebo group. Moreover, the risk of developing GC was lowered in 73% of participants who were confirmed for H. pylori ablation than those who had persistent infection of the bacteria. These results confirm the potential use of RFs as therapeutic targets for cancer therapy.
The H. pylori bacteria initiates GC by causing the DNA to replicate faster due to the chronic inflammation incited by the organism, and this leads to mutagenesis and genomic instability. Inflammation is a hallmark of cancer which plays a key role in all three carcinogenesis stages [52]. The virulence factors of H. pylori, such as the cytotoxin-associated gene A (cagA), are responsible for its chronic inflammation properties. CagA functions as an oncoprotein and can trigger MAPK signaling of host cells, leading to persistent inflammation and uncontrollable proliferation [44]. Additionally, the MAPK pathway is responsible for chemoresistance in pancreatic cancer and GC cells [53][54]. It is postulated that cagA travels through the type IV secretion systems (T4SS) upon contact with the host cell and this triggers the endocytosis of the protein (Figure 2) [55]. The protein can activate the MAPK/ERK pathway in two ways: by direct binding in a phosphorylation-independent state or through recruiting the phosphatase SHP2 [56][57]. The SHP2 protein plays a crucial role in the pathologic activity of cagA and can independently modify ERK signals autonomous of Ras [58].
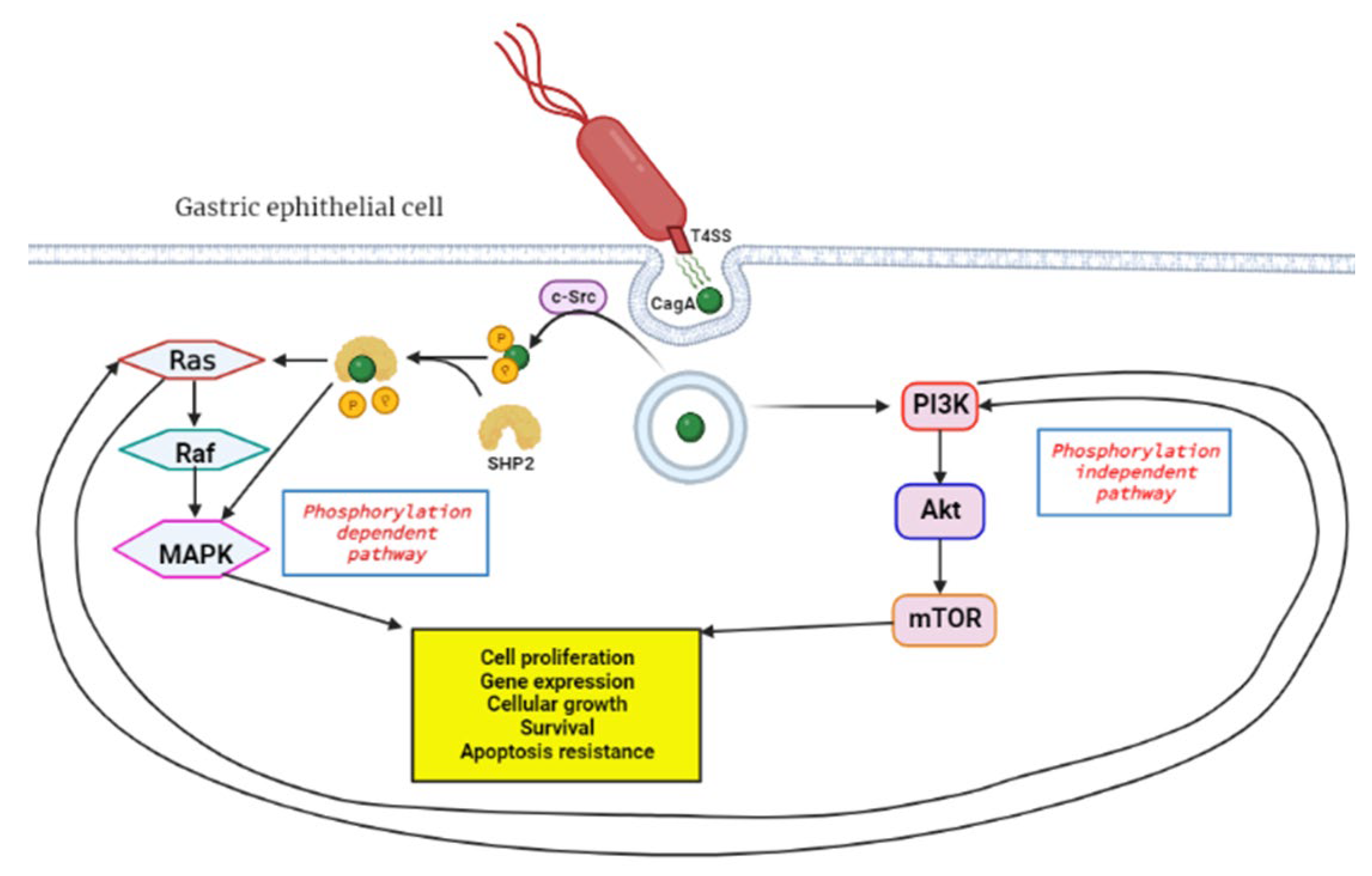
Figure 2. Activation of RAS/RAF/MEK/ERK pathway by H. pylori cagA oncoprotein. Upon contact with the gastric epithelial cell membrane, the bacteria’s T4SS system releases cagA through a channel, and this triggers endocytosis, a process where proteins get engulfed into the cell. In the phosphorylation dependent pathway c-Src, tyrosine kinase phosphorylates cagA, followed by SHP2 phosphatase cleavage of the phosphate groups from cagA. This leads to downstream activation of the RAS/RAF/MEK/ERK signal transduction pathway which favors tumorigenesis. The phosphorylation independent pathway is the PI3K/Akt/mTOR, which gets activated by cagA and results in products that induce tumorigenesis. Created with BioRender.com. (accessed on 27 September 2022).
The MAPK/ERK is also known as the RAS/RAF/MEK/ERK signaling pathway and plays a major role in regulating cell differentiation, proliferation and survival. This pathway is interlinked with the PI3K/Akt/mTOR pathway and can cause compensatory signal transduction in cases where the other is compromised [59]. The coupled inhibition of the two pathways has been effective in tumor stasis and overcoming drug resistance of GI tumor cells [60][61]. An elevated expression of the Ras protein is positively associated with increased Akt protein levels. Thus, PI3K/Akt/mTOR is an alternative pathway to Ras/Raf/MEK/ERK for EGFR signaling [62]. This may affect the efficacy of anticancer treatment, and therefore this must be considered when developing novel anticancer therapies. The bacteria can also initiate GC through aberrant DNA methylation, which will be discussed in more detail later in the review. Moreover, the expression of DNA mismatch repair (MMR) genes MutS and MutL are decreased in H- pylori-positive gastric mucosa [63].
The antibiotic metronidazole functions by interacting with the DNA of the target organisms (Gram-negative bacteria) breaking down DNA strands and causing the loss of DNA integrity and the ultimate inhibition of protein synthesis [64]. Metronidazole gets activated upon reduction by the protein ferredoxin (Figure 3). The concentration gradient created upon reduction increases the diffusion of metronidazole into the bacterial cell and cytotoxic free radical generation [64]. The drug has been shown to be successful in treating H. pylori; however, the bacteria has evolved to be resistant to it and can only be effective when in combination with esomeprazole and amoxicillin [65][66].
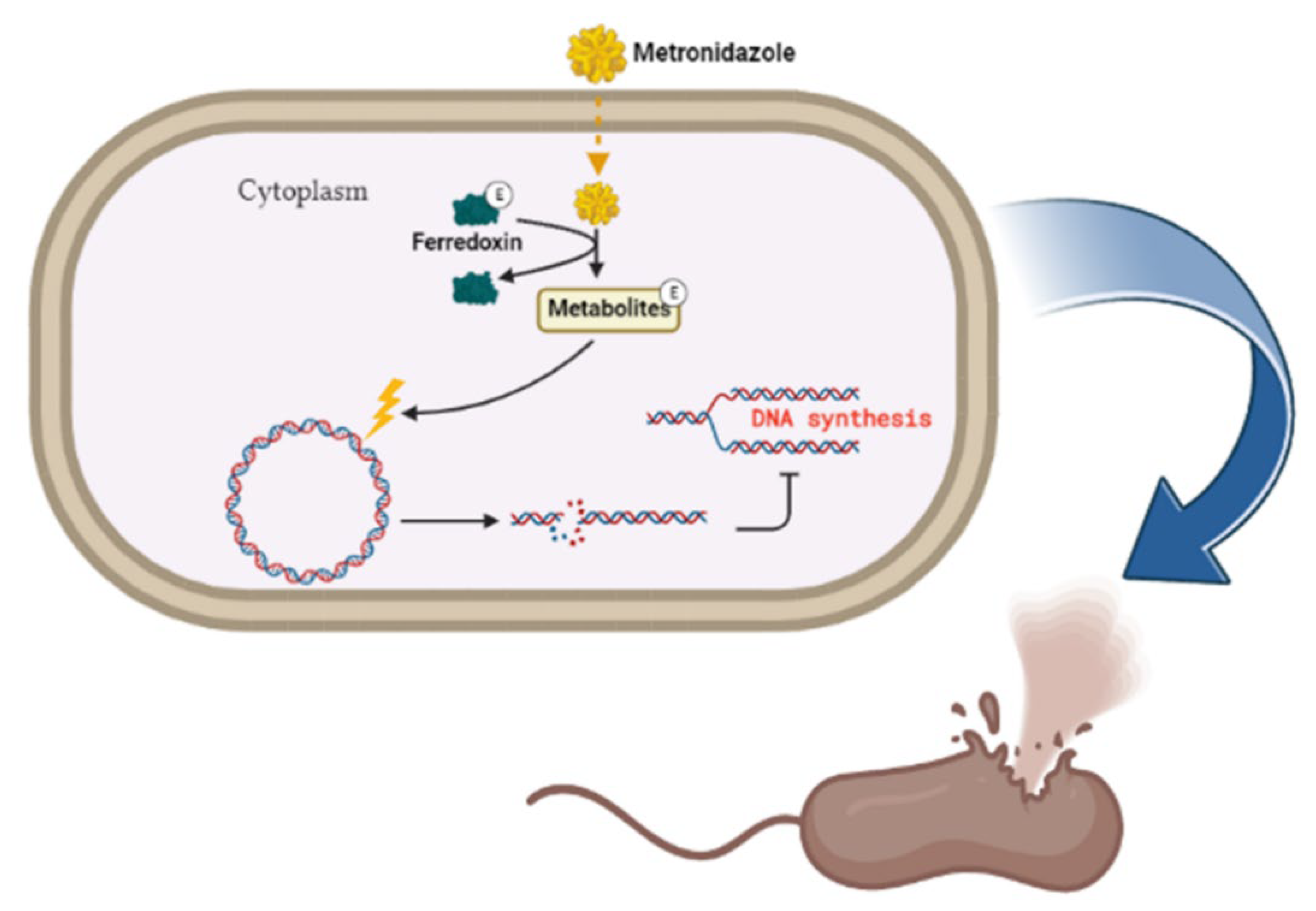
Figure 3. Metronidazole’s mode of action. The inert drug enters susceptible bacterial cells through passive diffusion. Metronidazole is activated through its reduction by ferredoxin. Upon activation of the drug, a concentration gradient is formed, and this favors the increased uptake of the drug into the organism, thus elevating its antimicrobial effect. DNA damage subsequently leads to protein synthesis inhibition and consequent apoptosis. Created with BioRender.com. (accessed on 27 September 2022).
Another type of Gram-negative bacteria, Fusobacteria, is considered a RF for GC. The Fusobacterium spp., predominantly F. nucleatum, are frequently found in abundance in GC, pancreatic and colorectal tumors compared to non-cancerous tissues [17][67][68]. F. nucleatum-positivity has been linked to overall worse survival in Lauren’s diffuse type of GC and MSI-high status of colon cancer [67][69]. It is therefore safe to assume that F. nucleatum predisposes individuals to the MSI-high subtype of GC. Just like H. pylori, Fusobacteria can be eradicated with metronidazole therapy. The bacteria is known to be highly sensitive to the drug [70]. Apart from bacteria, other viruses such as Epstein-Barr virus (EBV), which is sometimes referred to as human herpesvirus 4 (HHV4), raises the risk of GC by a factor of 18 times and the EBV-associated GC (EBVaGC) is observed more in males than in females [71]. EBVaGC contributes to approximately 10% of all GC cases worldwide and is more common in the early stages of the disease [72]. Other viruses with potential association with GC include the human papillomavirus (HPV), hepatitis B virus (HBV), John Cummingham virus (human polyomavirus 2) and human cytomegalovirus [73]. More research on these viruses is required to determine their role in GC pathogenesis.
2. Other Microbes Implicated in GC Pathogenesis
Carcinogenesis describes the process of cancer formation which stems from irreversible genetic alterations or interruptions due to internal and external factors. It is a multistage molecular process involving (i) initiation, (ii) promotion and (iii) progression [74][75]. The microbes can either directly affect the cells and lead to carcinogenesis or tamper with the body’s cellular pathways to support its growth and sustainability. The gut microbiome, which is also known as the human second genome, plays a major role in the pathogenesis of GI cancers including colorectal, pancreatic, liver and gastric [17][76][77][78]. Bacterial and viral pathogens negatively influence the host’s genomic stability and integrity by the destruction of DNA strands, thereby initiating tumor development [79]. Approximately 95% of the human body’s microbiota resides in the gut and the microbes generally assist in maintaining the balance between health and disease [80]. There appears to be microbiome dysbiosis in most cancers, and this has been found to aggravate tumorigenesis. Although the microbiome is implicated in a number of cancers, the exact mechanisms by which they lead to cancer is still controversial. This is due to the low biomass of the microbiota in the TME, making it challenging to study them further [81]. Thanks to omics studies, this challenge can be overcome, as they shed light on the role of the gut microbiome in cancer pathology, prevention, and therapy [82].
2.1. The Boas-Oppler Bacillus
The lactic acid bacillus (lactobacillus), which is commonly called the Boas-Oppler Bacillus, dates back to 1895 when Izmar Isidor Boas and Bruno Oppler described the role of these Gram-positive bacteria in GC [83]. In their study, the researchers discovered that the bacillus was present in abundance in the gastric juices of 95% of GC individuals included in the study. This has been observed to be common, especially in patients with an advanced stage of the disease [84]. Lertpiriyapong et al. reported that infection of insulin–gastrin (INS-GAS) transgenic mice with L. murinus ASF361 led to the development of gastric neoplasia via the upregulation of oncogenes and pro-inflammatory genes [85]. Lactobacilli produce lactic acid/lactate which plays a huge role in the Warburg effect, a hallmark of cancer. Additionally, the Lactobacilli play a role in the production of excessive amounts of N-nitroso compounds (NOCs), which are carcinogenic and predispose H. pylori-free individuals to GC [86][87]
2.2. Mycoplasma
The study of the role of mycoplasma infection in cancer development dates way back to the 1950s [88]. They are Gram-negative bacteria that belong to the class Mollicutes [89]. The bacteria are commonly known for causing infections of the ear, respiratory system, lungs, urogenital tract and also to cause sexually transmitted infections (STIs) [90]. The well-studied pathogenic species include Ureaplasma urealyticum, M. fermentans, M. penetrans, M. hominis, M. genitalium, M. pneumoniae, M. hyorhinis and os on. The M. hyorhinis species is implicated in the development of GC [88][91][92][93][94]. Although mycoplasma have been detected in GC biopsies, the infection is not considered a RF for the disease [95]. Research has shown that mycoplasma cause inflammation, which instigates cancer initiation and progression [96][97].
A p37 lipoprotein located on the outer membrane of M. hyorhinis has been proven to play a key role in tumorigenesis [93][98][99]. The p37 protein heightens the expression of inflammation-associated genes such as vascular cell adhesion molecule 1 (Vcam1), IL-6, IL-1, and lipocalin 2 (LCN2) [96]. Additionally, p37 promotes cell invasiveness by blocking contact inhibition, and this has been observed in melanoma, gastric and prostate carcinomas [93][98][99]. Gong et al. demonstrated that p37 promotes the metastasis of human GC and lung cancer cells through the activation of matrix metalloproteinase-2 (MMP-2) and EGFR/PI3K/AKT/ERK pathways [91]. Another mechanism by which the mycoplasma promotes metastasis is via the accumulation of β-catenin and the activation of its Wnt signaling pathway [98][100]. Moreover, metastasis in GC by M. hyorhinis can be initiated through activation of the NACHT, LRR, and PYD domains-containing protein 3 (NLRP3) [101]. The NLRP3 is an inflammasome critical in caspase-1 modulated inflammation in response to pathogenic organisms [102]. Because these organisms lack peptidoglycan and are Gram-negative, this makes them extremely resistant to antibiotics [90]. The M. hyorhinis infection has been linked with the diffuse-type GC with a higher infection rate in advanced stages (TNM III/IV) than in earlier stages of the disease [94]. On the other hand, M. hyorhinis can cause chronic infections which induce chromosomal instability, and one can classify this under the TCGA CIN subtype of GC [7][101]. The age group of GC patients who are more likely to be infected with mycoplasma is the elderly [94].
3. Compounds Linked with GC Induction
3.1. Contribution of Microbes in Asbestos-Induced GC
Amosite, actinolite, chrysotile, anthophyllite, crocidolite, and tremolite are the six types of asbestos of which chrysotile (white asbestos) is the most abundant (99%) and is also exceedingly hazardous and lethal [103][104]. However, this does not mean that the other types are less harmful, as they also possess toxicity to some extent [105]. The link between asbestos and GI cancers was first demonstrated by Selikoff et al. in 1960, then in 2012, a review by Kim et al. reported that among all GI cancers, GC is the one that is greatly linked with asbestos exposure [106][107]. Oksa et al. summarized the association between GC and asbestos exposure and concluded that the risk of developing GC is directly proportional to asbestos exposure with the risk ranging from 15% to 20% [108]. In a study by Patel-Mandlik and Millette, an olive baboon that was fed chrysotile was discovered to have asbestos fibers deposited in its stomach while other pieces were able to relocate to most neighboring tissues except for the small intestine [109]. This shows that the fibers cannot be digested and can remain in the stomach for longer periods of time before their excretion [110]. Because of its strength and chemical properties, the material does not get digested or broken down, and the exposure elicits scarring and irritation, resulting in inflammation of the tissue [110]. Data shows that prolonged asbestos exposure leads to chronic inflammation and cellular stress, which activates the MAPK pathway and related transcription factors leading to immune response gene expression [111]. The gut microbiome plays a vital role in modulating immune homeostasis and GC inflammation. However, its association with asbestos in cancer is poorly reported.
Stanik et al. evaluated the ability of L. casei and L. plantarum to biologically break down white asbestos fibers [112]. The bacteria were successful due to their ability to produce lactic acid, which contains hydrogen ions that can remove magnesium ions from the crystalline structure of the asbestos fibers. A study by Seshan showed that when chrysotile is exposed to strong acids like those of the stomach or water, the physical and chemical properties of the asbestos change as the magnesium is lost from the asbestos [113]. There is evidence that shows that L. plantarum is capable of preventing H. pylori-induced inflammation of the gastric mucosa and restores balance to the gut microbiome, which is altered during such an infection [114]. Pretreatment with these bacteria was able to slow down the expression of inflammatory cytokines and cell infiltration. Similarly, L. casei has an anti-cancer effect, as it is able to inhibit the mTOR and NF-kB signaling pathways, thereby leading to the cellular apoptosis of GC cells [115]. Figure 4 shows how L. casei and L. plantarum could potentially prevent GC carcinogenesis. From this information, it could be deduced that these two Lactobacilli species can be used to aid in the treatment of GC, more in particular one with an asbestos origin.
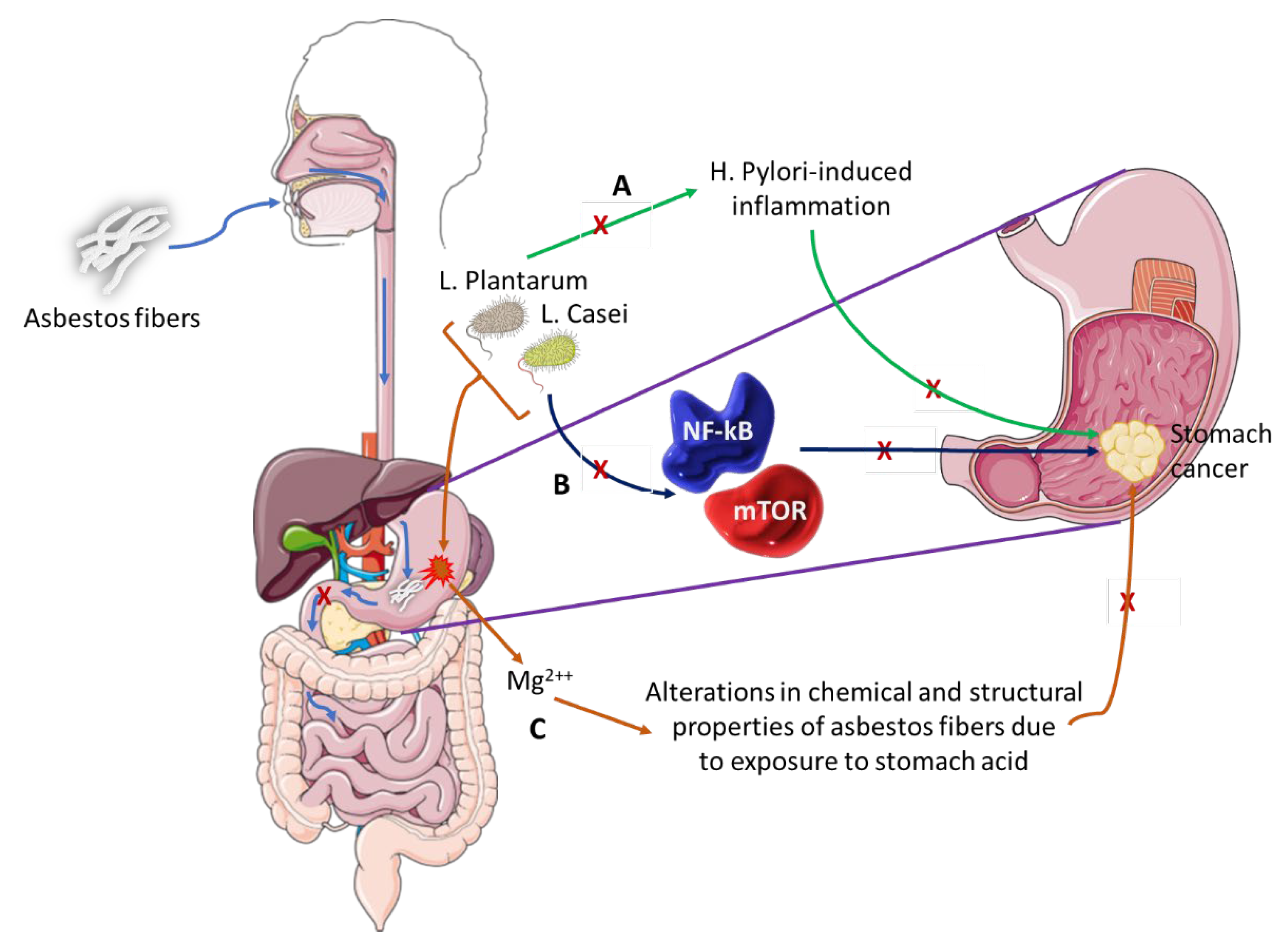
Figure 4. The role of microbiota in asbestos-induced GC.
Asbestos fibers can be ingested and pass through the esophagus and lodge into the stomach lining. These fibers do not pass through to the small intestines where they could possibly go through the process of excretion but remain in the stomach long enough to induce GC.
- (A)
-
L. Plantarum has the ability to block H. Pylori-induced inflammation that is associated with GC development.
- (B)
-
L. Casei bacterium downregulates pro-oncogenic signaling pathways (NF-kB and mTOR) thus inhibiting cancer development and progression.
- (C)
-
This pair of bacteria can alter the chemical and structural properties of white asbestos by the removal of magnesium ions, a process that could be explored as preventative therapy in individuals exposed to asbestos fibers or as therapeutic intervention in asbestos-induced GC.
3.2. Enterobacteriaceae and Nitrosamines Production
Nitrosamines are carcinogenic N-nitroso compounds which can nest in the stomach and are produced when amines react with nitrites. They can either be ingested as an outside source or produced from ingested food with the help of certain bacteria [116]. Foods that contain nitrites include processed meats, fish, fried bacon, beverages, and cheese [116]. Cigarettes and E-cigars also release some nitrosamines called tobacco-specific nitrosamines (TSNAs) when inhaled and can result in DNA damage and mutagenesis [117]. Exposure has been correlated with GC RFs such as diabetes and pathogenesis to the mammary glands, leading to breast cancer [118][119][120]. In breast cancer TSNAs actively bind nicotinic acetylcholine receptors (nAChRs), activating its signaling pathways. The alpha7 receptor (α7nAChR) is an oncoprotein that plays a role in both the initiation and progression stages of breast cancer carcinogenesis [121]. The estrogen receptor-positive type of breast cancer carcinoma has been shown to express the α7nAChR in high levels [121]. Nitrosamines have been reported in lung cancer as activators of the NF-kB and PI3K/AKT signaling pathways, which are pivotal in cell proliferation [122].
There is a positive correlation between ingestion from nitrosamine sources and GC [123][124][125]. The nitrosamine hypothesis dates back to the 1950s and paved the way for research investigating the role of the gut microbiome and GC until the focus shifted towards H. pylori’s role in chronic inflammation. Nitrate reductases are secreted by Gram-negative bacteria called Enterobacteriaceae. These enzymes catalyze the conversion of nitrate to nitrite [126]. These bacteria play a key role in nitrosamine production in the gut. In a study by Sarhadi et al., Enterobacteriaceae was found to be abundant in fecal samples of different GC types [127]. Similar findings were reported by Liu et al., who detected Escherichia and Streptococcaceae in abundance in GC patients [128]. Qin et al. reported an abundance of Enterobacteriaceae in diabetic patients, one of the risk factors of GC [129].
References
- Yusefi, A.R.; Lankarani, K.B.; Bastani, P.; Radinmanesh, M.; Kavosi, Z. Risk factors for gastric cancer: A systematic review. Asian Pac. J. Cancer Prev. APJCP 2018, 19, 591.
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424.
- Smyth, E.C.; Nilsson, M.; Grabsch, H.I.; van Grieken, N.C.; Lordick, F. Gastric cancer. Lancet 2020, 396, 635–648.
- Hooi, J.K.; Lai, W.Y.; Ng, W.K.; Suen, M.M.; Underwood, F.E.; Tanyingoh, D.; Malfertheiner, P.; Graham, D.Y.; Wong, V.W.; Wu, J.C. Global prevalence of Helicobacter pylori infection: Systematic review and meta-analysis. Gastroenterology 2017, 153, 420–429.
- Li, F.; Du, H.; Li, S.; Liu, J. The association between metabolic syndrome and gastric cancer in Chinese. Front. Oncol. 2018, 8, 326.
- Tsugane, S.; Sasazuki, S. Diet and the risk of gastric cancer: Review of epidemiological evidence. Gastric Cancer 2007, 10, 75–83.
- Network, C.G.A.R. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202.
- Kusano, M.; Toyota, M.; Suzuki, H.; Akino, K.; Aoki, F.; Fujita, M.; Hosokawa, M.; Shinomura, Y.; Imai, K.; Tokino, T. Genetic, epigenetic, and clinicopathologic features of gastric carcinomas with the CpG island methylator phenotype and an association with Epstein–Barr virus. Cancer 2006, 106, 1467–1479.
- Loh, M.; Liem, N.; Vaithilingam, A.; Lim, P.L.; Sapari, N.S.; Elahi, E.; Mok, Z.Y.; Cheng, C.L.; Yan, B.; Pang, B. DNA methylation subgroups and the CpG island methylator phenotype in gastric cancer: A comprehensive profiling approach. BMC Gastroenterol. 2014, 14, 55.
- Arnold, M.; Park, J.Y.; Camargo, M.C.; Lunet, N.; Forman, D.; Soerjomataram, I. Is gastric cancer becoming a rare disease? A global assessment of predicted incidence trends to 2035. Gut 2020, 69, 823–829.
- Heer, E.V.; Harper, A.S.; Sung, H.; Jemal, A.; Fidler-Benaoudia, M.M. Emerging cancer incidence trends in Canada: The growing burden of young adult cancers. Cancer 2020, 126, 4553–4562.
- Alegría-Torres, J.A.; Baccarelli, A.; Bollati, V. Epigenetics and lifestyle. Epigenomics 2011, 3, 267–277.
- Lauby-Secretan, B.; Scoccianti, C.; Loomis, D.; Grosse, Y.; Bianchini, F.; Straif, K. Body fatness and cancer—Viewpoint of the IARC Working Group. N. Engl. J. Med. 2016, 375, 794–798.
- Haydon, A.M.; MacInnis, R.J.; English, D.R.; Giles, G.G. Effect of physical activity and body size on survival after diagnosis with colorectal cancer. Gut 2006, 55, 62–67.
- Renehan, A.G.; Soerjomataram, I.; Tyson, M.; Egger, M.; Zwahlen, M.; Coebergh, J.W.; Buchan, I. Incident cancer burden attributable to excess body mass index in 30 European countries. Int. J. Cancer 2010, 126, 692–702.
- Ley, R.E.; Bäckhed, F.; Turnbaugh, P.; Lozupone, C.A.; Knight, R.D.; Gordon, J.I. Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. USA 2005, 102, 11070–11075.
- Wei, M.-Y.; Shi, S.; Liang, C.; Meng, Q.-C.; Hua, J.; Zhang, Y.-Y.; Liu, J.; Zhang, B.; Xu, J.; Yu, X.-J. The microbiota and microbiome in pancreatic cancer: More influential than expected. Mol. Cancer 2019, 18, 97.
- Kim, M.-H.; Yun, K.E.; Kim, J.; Park, E.; Chang, Y.; Ryu, S.; Kim, H.-L.; Kim, H.-N. Gut microbiota and metabolic health among overweight and obese individuals. Sci. Rep. 2020, 10, 19417.
- Tseng, C.-H.; Tseng, F.-H. Diabetes and gastric cancer: The potential links. World J. Gastroenterol. WJG 2014, 20, 1701.
- Dabo, B.; Pelucchi, C.; Rota, M.; Jain, H.; Bertuccio, P.; Bonzi, R.; Palli, D.; Ferraroni, M.; Zhang, Z.-F.; Sanchez-Anguiano, A. The association between diabetes and gastric cancer: Results from the Stomach Cancer Pooling Project Consortium. Eur. J. Cancer Prev. 2022, 31, 260.
- Derakhshan, M.H.; Malekzadeh, R.; Watabe, H.; Yazdanbod, A.; Fyfe, V.; Kazemi, A.; Rakhshani, N.; Didevar, R.; Sotoudeh, M.; Zolfeghari, A. Combination of gastric atrophy, reflux symptoms and histological subtype indicates two distinct aetiologies of gastric cardia cancer. Gut 2008, 57, 298–305.
- Rubenstein, J.H.; Taylor, J. Meta-analysis: The association of oesophageal adenocarcinoma with symptoms of gastro-oesophageal reflux. Aliment. Pharmacol. Ther. 2010, 32, 1222–1227.
- Wu, A.H.; Tseng, C.C.; Bernstein, L. Hiatal hernia, reflux symptoms, body size, and risk of esophageal and gastric adenocarcinoma. Cancer Interdiscip. Int. J. Am. Cancer Soc. 2003, 98, 940–948.
- Ye, W.; Chow, W.-H.; Lagergren, J.; Yin, L.; Nyrén, O. Risk of adenocarcinomas of the esophagus and gastric cardia in patients with gastroesophageal reflux diseases and after antireflux surgery. Gastroenterology 2001, 121, 1286–1293.
- Li, Y.; Feng, A.; Zheng, S.; Chen, C.; Lyu, J. Recent Estimates and Predictions of 5-Year Survival in Patients with Gastric Cancer: A Model-Based Period Analysis. Cancer Control 2022, 29, 10732748221099227.
- Mukaisho, K.-i.; Nakayama, T.; Hagiwara, T.; Hattori, T.; Sugihara, H. Two distinct etiologies of gastric cardia adenocarcinoma: Interactions among pH, Helicobacter pylori, and bile acids. Front. Microbiol. 2015, 6, 412.
- Misumi, A.; Murakami, A.; Harada, K.; Baba, K.; Akagi, M. Definition of carcinoma of the gastric cardia. Langenbecks Arch. Chir. 1989, 374, 221–226.
- Alkhathami, A.M.; Alzahrani, A.A.; Alzhrani, M.A.; Alsuwat, O.B.; Mahfouz, M.E.M. Risk factors for gastroesophageal reflux disease in Saudi Arabia. Gastroenterol. Res. 2017, 10, 294–300.
- Naito, Y.; Kashiwagi, K.; Takagi, T.; Andoh, A.; Inoue, R. Intestinal dysbiosis secondary to proton-pump inhibitor use. Digestion 2018, 97, 195–204.
- Polat, F.R.; Polat, S. The effect of Helicobacter pylori on gastroesophageal reflux disease. JSLS J. Soc. Laparoendosc. Surg. 2012, 16, 260.
- Argilés, J.M.; López-Soriano, F.J. The role of cytokines in cancer cachexia. Med. Res. Rev. 1999, 19, 223–248.
- Germano, G.; Allavena, P.; Mantovani, A. Cytokines as a key component of cancer-related inflammation. Cytokine 2008, 43, 374–379.
- Nicolini, A.; Carpi, A.; Rossi, G. Cytokines in breast cancer. Cytokine Growth Factor Rev. 2006, 17, 325–337.
- Baj, J.; Forma, A.; Sitarz, M.; Portincasa, P.; Garruti, G.; Krasowska, D.; Maciejewski, R. Helicobacter pylori virulence factors—mechanisms of bacterial pathogenicity in the gastric microenvironment. Cells 2020, 10, 27.
- Ding, S.-Z.; Goldberg, J.B.; Hatakeyama, M. Helicobacter pylori infection, oncogenic pathways and epigenetic mechanisms in gastric carcinogenesis. Future Oncol. 2010, 6, 851–862.
- Pai, S.G.; Carneiro, B.A.; Mota, J.M.; Costa, R.; Leite, C.A.; Barroso-Sousa, R.; Kaplan, J.B.; Chae, Y.K.; Giles, F.J. Wnt/beta-catenin pathway: Modulating anticancer immune response. J. Hematol. Oncol. 2017, 10, 101.
- Cui, J.; Jiang, W.; Wang, S.; Wang, L.; Xie, K. Role of Wnt/β-catenin signaling in drug resistance of pancreatic cancer. Curr. Pharm. Des. 2012, 18, 2464–2471.
- Mitchell, D.R.; Derakhshan, M.H.; Wirz, A.A.; Orange, C.; Ballantyne, S.A.; Going, J.J.; McColl, K.E. The gastric acid pocket is attenuated in H. pylori infected subjects. Gut 2017, 66, 1555–1562.
- Hansson, L.-E.; Nyrén, O.; Hsing, A.W.; Bergström, R.; Josefsson, S.; Chow, W.-H.; Fraumeni, J.F., Jr.; Adami, H.-O. The risk of stomach cancer in patients with gastric or duodenal ulcer disease. N. Engl. J. Med. 1996, 335, 242–249.
- Mohammadi, S.O.; Yadegar, A.; Kargar, M.; Mirjalali, H.; Kafilzadeh, F. The impact of Helicobacter pylori infection on gut microbiota-endocrine system axis; modulation of metabolic hormone levels and energy homeostasis. J. Diabetes Metab. Disord. 2020, 19, 1855–1861.
- Bauerfeind, P.; Garner, R.; Dunn, B.; Mobley, H. Synthesis and activity of Helicobacter pylori urease and catalase at low pH. Gut 1997, 40, 25–30.
- Ahmed, A.; Smoot, D.; Littleton, G.; Tackey, R.; Walters, C.S.; Kashanchi, F.; Allen, C.R.; Ashktorab, H. Helicobacter pylori inhibits gastric cell cycle progression. Microbes Infect. 2000, 2, 1159–1169.
- Koeppel, M.; Garcia-Alcalde, F.; Glowinski, F.; Schlaermann, P.; Meyer, T.F. Helicobacter pylori infection causes characteristic DNA damage patterns in human cells. Cell Rep. 2015, 11, 1703–1713.
- Ding, S.Z.; Smith, M.F., Jr.; Goldberg, J.B. Helicobacter pylori and mitogen-activated protein kinases regulate the cell cycle, proliferation and apoptosis in gastric epithelial cells. J. Gastroenterol. Hepatol. 2008, 23, e67–e78.
- Rossi, C.; Cicalini, I.; Cufaro, M.C.; Consalvo, A.; Upadhyaya, P.; Sala, G.; Antonucci, I.; Del Boccio, P.; Stuppia, L.; De Laurenzi, V. Breast cancer in the era of integrating “Omics” approaches. Oncogenesis 2022, 11, 17.
- Goodman, A.M.; Holden, K.A.; Jeong, A.-R.; Kim, L.; Fitzgerald, K.D.; Almasri, E.; McLennan, G.; Eisenberg, M.; Jahromi, A.H.; Hoh, C. Response to CAR-T Therapy Can be Monitored Using Genome-Wide Sequencing of Cell-Free DNA in Patients with DLBCL. Blood 2020, 136, 17.
- Goonetilleke, K.; Siriwardena, A. Systematic review of carbohydrate antigen (CA 19-9) as a biochemical marker in the diagnosis of pancreatic cancer. Eur. J. Surg. Oncol. (EJSO) 2007, 33, 266–270.
- Ray, K. Biomarkers for the early detection of PDAC. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 505.
- Breijyeh, Z.; Jubeh, B.; Karaman, R. Resistance of gram-negative bacteria to current antibacterial agents and approaches to resolve it. Molecules 2020, 25, 1340.
- Choi, I.J.; Kook, M.-C.; Kim, Y.-I.; Cho, S.-J.; Lee, J.Y.; Kim, C.G.; Park, B.; Nam, B.-H. Helicobacter pylori therapy for the prevention of metachronous gastric cancer. N. Engl. J. Med. 2018, 378, 1085–1095.
- Choi, I.J.; Kim, C.G.; Lee, J.Y.; Kim, Y.-I.; Kook, M.-C.; Park, B.; Joo, J. Family history of gastric cancer and Helicobacter pylori treatment. N. Engl. J. Med. 2020, 382, 427–436.
- Pappas-Gogos, G.; Tepelenis, K.; Fousekis, F.; Katsanos, K.; Pitiakoudis, M.; Vlachos, K. The Implication of Gastric Microbiome in the Treatment of Gastric Cancer. Cancers 2022, 14, 2039.
- Guo, X.; Ma, N.; Wang, J.; Song, J.; Bu, X.; Cheng, Y.; Sun, K.; Xiong, H.; Jiang, G.; Zhang, B. Increased p38-MAPK is responsible for chemotherapy resistance in human gastric cancer cells. BMC Cancer 2008, 8, 375.
- Zhao, Y.; Shen, S.; Guo, J.; Chen, H.; Greenblatt, D.Y.; Kleeff, J.; Liao, Q.; Chen, G.; Friess, H.; Leung, P.S. Mitogen-activated protein kinases and chemoresistance in pancreatic cancer cells. J. Surg. Res. 2006, 136, 325–335.
- Tohidpour, A.; Gorrell, R.J.; Roujeinikova, A.; Kwok, T. The middle fragment of Helicobacter pylori CagA induces actin rearrangement and triggers its own uptake into gastric epithelial cells. Toxins 2017, 9, 237.
- Krisch, L.M.; Posselt, G.; Hammerl, P.; Wessler, S. CagA phosphorylation in Helicobacter pylori-infected B cells is mediated by the nonreceptor tyrosine kinases of the Src and Abl families. Infect. Immun. 2016, 84, 2671–2680.
- Selbach, M.; Paul, F.E.; Brandt, S.; Guye, P.; Daumke, O.; Backert, S.; Dehio, C.; Mann, M. Host cell interactome of tyrosine-phosphorylated bacterial proteins. Cell Host Microbe 2009, 5, 397–403.
- Higashi, H.; Nakaya, A.; Tsutsumi, R.; Yokoyama, K.; Fujii, Y.; Ishikawa, S.; Higuchi, M.; Takahashi, A.; Kurashima, Y.; Teishikata, Y. Helicobacter pylori CagA induces Ras-independent morphogenetic response through SHP-2 recruitment and activation. J. Biol. Chem. 2004, 279, 17205–17216.
- Shimizu, T.; Tolcher, A.W.; Papadopoulos, K.P.; Beeram, M.; Rasco, D.W.; Smith, L.S.; Gunn, S.; Smetzer, L.; Mays, T.A.; Kaiser, B. The Clinical Effect of the Dual-Targeting Strategy Involving PI3K/AKT/mTOR and RAS/MEK/ERK Pathways in Patients with Advanced CancerClinical Effect of Dual PI3K and MAPK Pathways Inhibitions. Clin. Cancer Res. 2012, 18, 2316–2325.
- Gupta, A.; Ma, S.; Che, K.; Pobbati, A.V.; Rubin, B.P. Inhibition of PI3K and MAPK pathways along with KIT inhibitors as a strategy to overcome drug resistance in gastrointestinal stromal tumors. PLoS ONE 2021, 16, e0252689.
- Roberts, P.J.; Usary, J.E.; Darr, D.B.; Dillon, P.M.; Pfefferle, A.D.; Whittle, M.C.; Duncan, J.S.; Johnson, S.M.; Combest, A.J.; Jin, J. Combined PI3K/mTOR and MEK Inhibition Provides Broad Antitumor Activity in Faithful Murine Cancer ModelsCombined PI3K/mTOR and MEK Inhibition. Clin. Cancer Res. 2012, 18, 5290–5303.
- Khwanraj, K.; Madlah, S.; Grataitong, K.; Dharmasaroja, P. Comparative mRNA expression of eEF1A isoforms and a PI3K/Akt/mTOR pathway in a cellular model of Parkinson’s disease. Parkinson’s Dis. 2016, 2016, 8716016.
- Kim, J.J.; Tao, H.; Carloni, E.; Leung, W.K.; Graham, D.Y.; Sepulveda, A.R. Helicobacter pylori impairs DNA mismatch repair in gastric epithelial cells. Gastroenterology 2002, 123, 542–553.
- Weir, C.B.; Le, J.K. Metronidazole. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021.
- Adachi, T.; Matsui, S.; Watanabe, T.; Okamoto, K.; Okamoto, A.; Kono, M.; Yamada, M.; Nagai, T.; Komeda, Y.; Minaga, K. Comparative study of clarithromycin-versus metronidazole-based triple therapy as first-line eradication for Helicobacter pylori. Oncology 2017, 93, 15–19.
- Jenks, P.J.; Edwards, D.I. Metronidazole resistance in Helicobacter pylori. Int. J. Antimicrob. Agents 2002, 19, 1–7.
- Boehm, E.T.; Thon, C.; Kupcinskas, J.; Steponaitiene, R.; Skieceviciene, J.; Canbay, A.; Malfertheiner, P.; Link, A. Fusobacterium nucleatum is associated with worse prognosis in Lauren’s diffuse type gastric cancer patients. Sci. Rep. 2020, 10, 16240.
- Mima, K.; Nishihara, R.; Qian, Z.R.; Cao, Y.; Sukawa, Y.; Nowak, J.A.; Yang, J.; Dou, R.; Masugi, Y.; Song, M. Fusobacterium nucleatum in colorectal carcinoma tissue and patient prognosis. Gut 2016, 65, 1973–1980.
- Oh, H.J.; Kim, J.H.; Bae, J.M.; Kim, H.J.; Cho, N.-Y.; Kang, G.H. Prognostic impact of Fusobacterium nucleatum depends on combined tumor location and microsatellite instability status in stage II/III colorectal cancers treated with adjuvant chemotherapy. J. Pathol. Transl. Med. 2019, 53, 40–49.
- Kim, M.; Yun, S.Y.; Lee, Y.; Lee, H.; Yong, D.; Lee, K. Clinical differences in patients infected with fusobacterium and antimicrobial susceptibility of fusobacterium isolates recovered at a tertiary-care hospital in korea. Ann. Lab. Med. 2022, 42, 188–195.
- Tavakoli, A.; Monavari, S.H.; Solaymani Mohammadi, F.; Kiani, S.J.; Armat, S.; Farahmand, M. Association between Epstein-Barr virus infection and gastric cancer: A systematic review and meta-analysis. BMC Cancer 2020, 20, 493.
- Iizasa, H.; Nanbo, A.; Nishikawa, J.; Jinushi, M.; Yoshiyama, H. Epstein-Barr Virus (EBV)-associated gastric carcinoma. Viruses 2012, 4, 3420–3439.
- Wang, H.; Chen, X.-L.; Liu, K.; Bai, D.; Zhang, W.-H.; Chen, X.-Z.; Hu, J.-K. Associations between gastric cancer risk and virus infection other than Epstein-Barr virus: A systematic review and meta-analysis based on epidemiological studies. Clin. Transl. Gastroenterol. 2020, 11, e00201.
- Malarkey, D.E.; Hoenerhoff, M.J.; Maronpot, R.R. Carcinogenesis: Manifestation and mechanisms. Fundam. Toxicol. Pathol. 2018, 83–104.
- Pitot, H.C. The molecular biology of carcinogenesis. Cancer 1993, 72, 962–970.
- Elaskandrany, M.; Patel, R.; Patel, M.; Miller, G.; Saxena, D.; Saxena, A. Fungi, host immune response, and tumorigenesis. Am. J. Physiol. -Gastrointest. Liver Physiol. 2021, 321, G213–G222.
- Kaźmierczak-Siedlecka, K.; Dvořák, A.; Folwarski, M.; Daca, A.; Przewłócka, K.; Makarewicz, W. Fungal gut microbiota dysbiosis and its role in colorectal, oral, and pancreatic carcinogenesis. Cancers 2020, 12, 1326.
- Zackular, J.P.; Baxter, N.T.; Iverson, K.D.; Sadler, W.D.; Petrosino, J.F.; Chen, G.Y.; Schloss, P.D. The gut microbiome modulates colon tumorigenesis. MBio 2013, 4, e00692-13.
- Geng, F.; Zhang, Y.; Lu, Z.; Zhang, S.; Pan, Y. Fusobacterium nucleatum caused DNA damage and promoted cell proliferation by the Ku70/p53 pathway in oral cancer cells. DNA Cell Biol. 2020, 39, 144–151.
- Cani, P.D. Human gut microbiome: Hopes, threats and promises. Gut 2018, 67, 1716–1725.
- Lee, M.H. Harness the functions of gut microbiome in tumorigenesis for cancer treatment. Cancer Commun. 2021, 41, 937–967.
- Knight, R.; Callewaert, C.; Marotz, C.; Hyde, E.R.; Debelius, J.W.; McDonald, D.; Sogin, M.L. The microbiome and human biology. Annu. Rev. Genom. Hum. Genet. 2017, 18, 65–86.
- Oppler, B. Zur Kenntniss des Mageninhalts beim Carcinoma ventriculi1. DMW-Dtsch. Med. Wochenschr. 1895, 21, 73–75.
- Galt, H.; Iles, C. A study of the Boas-Oppler bacillus. J. Pathol. Bacteriol. 1915, 19, 239–244.
- Lertpiriyapong, K.; Whary, M.T.; Muthupalani, S.; Lofgren, J.L.; Gamazon, E.R.; Feng, Y.; Ge, Z.; Wang, T.C.; Fox, J.G. Gastric colonisation with a restricted commensal microbiota replicates the promotion of neoplastic lesions by diverse intestinal microbiota in the Helicobacter pylori INS-GAS mouse model of gastric carcinogenesis. Gut 2014, 63, 54–63.
- Li, Z.-P.; Liu, J.-X.; Lu, L.-L.; Wang, L.-L.; Xu, L.; Guo, Z.-H.; Dong, Q.-J. Overgrowth of Lactobacillus in gastric cancer. World J. Gastrointest. Oncol. 2021, 13, 1099.
- Xu, L.; Qu, Y.-H.; Chu, X.-D.; Wang, R.; Nelson, H.H.; Gao, Y.-T.; Yuan, J.-M. Urinary levels of N-nitroso compounds in relation to risk of gastric cancer: Findings from the shanghai cohort study. PLoS ONE 2015, 10, e0117326.
- Butler, M.; Leach, R. 1116 Proceedings ofthe Royal Society ofMedicine 40. J. Bact 1956, 71, 362.
- Waites, K.B.; Katz, B.; Schelonka, R.L. Mycoplasmas and ureaplasmas as neonatal pathogens. Clin. Microbiol. Rev. 2005, 18, 757–789.
- Lanao, A.E.; Chakraborty, R.K.; Pearson-Shaver, A.L. Mycoplasma infections. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021.
- Gong, M.; Meng, L.; Jiang, B.; Zhang, J.; Yang, H.; Wu, J.; Shou, C. p37 from Mycoplasma hyorhinis promotes cancer cell invasiveness and metastasis through activation of MMP-2 and followed by phosphorylation of EGFR. Molecular cancer therapeutics 2008, 7, 530–537.
- Duan, H.; Qu, L.; Shou, C. Activation of EGFR-PI3K-AKT signaling is required for Mycoplasma hyorhinis-promoted gastric cancer cell migration. Cancer Cell Int. 2014, 14, 135.
- Ketcham, C.M.; Anai, S.; Reutzel, R.; Sheng, S.; Schuster, S.M.; Brenes, R.B.; Agbandje-McKenna, M.; McKenna, R.; Rosser, C.J.; Boehlein, S.K. p37 induces tumor invasiveness. Mol. Cancer Ther. 2005, 4, 1031–1038.
- Yang, H.; Qu, L.; Ma, H.; Chen, L.; Liu, W.; Liu, C.; Meng, L.; Wu, J.; Shou, C. Mycoplasma hyorhinisinfection in gastric carcinoma and its effects on the malignant phenotypes of gastric cancer cells. BMC Gastroenterol. 2010, 10, 132.
- Nascimento Araujo, C.d.; Amorim, A.T.; Barbosa, M.S.; Alexandre, J.C.P.L.; Campos, G.B.; Macedo, C.L.; Marques, L.M.; Timenetsky, J. Evaluating the presence of Mycoplasma hyorhinis, Fusobacterium nucleatum, and Helicobacter pylori in biopsies of patients with gastric cancer. Infect. Agents Cancer 2021, 16, 70.
- Gomersall, A.C.; Phan, H.A.; Iacuone, S.; Li, S.F.; Parish, R.W. The Mycoplasma hyorhinis p37 protein rapidly induces genes in fibroblasts associated with inflammation and cancer. PLoS ONE 2015, 10, e0140753.
- Patil, S.; Rao, R.S.; Raj, A.T. Role of Mycoplasma in the initiation and progression of oral cancer. J. Int. Oral Health JIOH 2015, 7, i–ii.
- Liu, W.-B.; Zhang, J.-Z.; Jiang, B.-H.; Ren, T.-T.; Gong, M.-M.; Meng, L.; Shou, C.-C. Lipoprotein p37 from Mycoplasma hyorhinis inhibiting mammalian cell adhesion. J. Biomed. Sci. 2006, 13, 323–331.
- Schmidhauser, C.; Dudler, R.; Schmidt, T.; Parish, R. A mycoplasmal protein influences tumour cell invasiveness and contact inhibition in vitro. J. Cell Sci. 1990, 95, 499–506.
- Takahashi-Yanaga, F.; Kahn, M. Targeting Wnt signaling: Can we safely eradicate cancer stem cells? Clin. Cancer Res. 2010, 16, 3153–3162.
- Xu, Y.; Li, H.; Chen, W.; Yao, X.; Xing, Y.; Wang, X.; Zhong, J.; Meng, G. Mycoplasma hyorhinis activates the NLRP3 inflammasome and promotes migration and invasion of gastric cancer cells. PLoS ONE 2013, 8, e77955.
- Guo, H.; Callaway, J.B.; Ting, J.P. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687.
- Zholobenko, V.; Rutten, F.; Zholobenko, A.; Holmes, A. In situ spectroscopic identification of the six types of asbestos. J. Hazard. Mater. 2021, 403, 123951.
- Landrigan, P.J.; Nicholson, W.J.; Suzuki, Y.; LaDou, J. The hazards of chrysotile asbestos: A critical review. Ind. Health 1999, 37, 271–280.
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Arsenic, Metals, Fibres, and Dusts. IARC Monogr. Eval. Carcinog. Risks Hum. 2012, 100, 11.
- Kim, S.J.; Williams, D.; Cheresh, P.; Kamp, D.W. Asbestos-induced gastrointestinal cancer: An update. J. Gastrointest. Dig. Syst. 2013, 3, 135.
- Selikoff, I.J.; Hammond, E.C.; Churg, J. Asbestos exposure, smoking, and neoplasia. JAMA 1968, 204, 106–112.
- Oksa, P.; Wolff, H.; Vehmas, T.; Pallasaho, P.; Frilander, H. Asbestos, Asbestosis, and Cancer: Helsinki Criteria for Diagnosis and Attribution 2014; Finnish Institute of Occupational Health: Helsinki, Finland, 2014.
- Patel-Mandlik, K.; Millette, J. Evidence of migration of ingested asbestos into various baboon organs. Scan. Electron Microsc. 1980, 1, 347–354.
- Luus, K. Asbestos: Mining exposure, health effects and policy implications. McGill J. Med. MJM 2007, 10, 121.
- Manning, C.B.; Vallyathan, V.; Mossman, B.T. Diseases caused by asbestos: Mechanisms of injury and disease development. Int. Immunopharmacol. 2002, 2, 191–200.
- Stanik, I.A.; Cedzynska, K.; Zakowska, Z. Destruction of the chrysotile asbestos structure with a population of bacteria Lactobacillus casei and Lactobacillus plantarum. Fresenius Environ. Bull. 2006, 15, 640.
- Seshan, K. How are the physical and chemical properties of chrysotile asbestos altered by a 10-year residence in water and up to 5 days in simulated stomach acid? Environ. Health Perspect. 1983, 53, 143–148.
- Pan, M.; Wan, C.; Xie, Q.; Huang, R.; Tao, X.; Shah, N.P.; Wei, H. Changes in gastric microbiota induced by Helicobacter pylori infection and preventive effects of Lactobacillus plantarum ZDY 2013 against such infection. J. Dairy Sci. 2016, 99, 970–981.
- Hwang, J.W.; Baek, Y.-M.; Yang, K.E.; Yoo, H.-S.; Cho, C.-K.; Lee, Y.-W.; Park, J.; Eom, C.-Y.; Lee, Z.-W.; Choi, J.-S. Lactobacillus casei extract induces apoptosis in gastric cancer by inhibiting NF-κB and mTOR-mediated signaling. Integr. Cancer Ther. 2013, 12, 165–173.
- Scanlan, R. Formation and occurrence of nitrosamines in food. Cancer Res. 1983, 43, 2435s–2440s.
- Kankanamage, R.N.; Ghosh, A.B.; Jiang, D.; Gkika, K.; Keyes, T.; Achola, L.A.; Suib, S.; Rusling, J.F. Metabolites of tobacco-and e-cigarette-related nitrosamines can drive Cu2+-mediated DNA oxidation. Chem. Res. Toxicol. 2020, 33, 2072–2086.
- de la Monte, S.M.; Neusner, A.; Chu, J.; Lawton, M. Epidemiological trends strongly suggest exposures as etiologic agents in the pathogenesis of sporadic Alzheimer’s disease, diabetes mellitus, and non-alcoholic steatohepatitis. J. Alzheimer’s Dis. 2009, 17, 519–529.
- Gankhuyag, N.; Lee, K.-H.; Cho, J.-Y. The role of nitrosamine (NNK) in breast cancer carcinogenesis. J. Mammary Gland. Biol. Neoplasia 2017, 22, 159–170.
- Tong, M.; Neusner, A.; Longato, L.; Lawton, M.; Wands, J.R.; de la Monte, S.M. Nitrosamine exposure causes insulin resistance diseases: Relevance to type 2 diabetes mellitus, non-alcoholic steatohepatitis, and Alzheimer’s disease. J. Alzheimer’s Dis. 2009, 17, 827–844.
- Lee, C.-H.; Chang, Y.-C.; Chen, C.-S.; Tu, S.-H.; Wang, Y.-J.; Chen, L.-C.; Chang, Y.-J.; Wei, P.-L.; Chang, H.-W.; Chang, C.-H. Crosstalk between nicotine and estrogen-induced estrogen receptor activation induces α9-nicotinic acetylcholine receptor expression in human breast cancer cells. Breast Cancer Res. Treat. 2011, 129, 331–345.
- Wen, J.; Fu, J.-H.; Zhang, W.; Guo, M. Lung carcinoma signaling pathways activated by smoking. Chin. J. Cancer 2011, 30, 551.
- Song, P.; Wu, L.; Guan, W. Dietary nitrates, nitrites, and nitrosamines intake and the risk of gastric cancer: A meta-analysis. Nutrients 2015, 7, 9872–9895.
- La Vecchia, C.; D’Avanzo, B.; Airoldi, L.; Braga, C.; Decarli, A. Nitrosamine intake and gastric cancer risk. Eur. J. Cancer Prev. 1995, 4, 469–474.
- Taneja, P.; Labhasetwar, P.; Nagarnaik, P.; Ensink, J.H. The risk of cancer as a result of elevated levels of nitrate in drinking water and vegetables in Central India. J. Water Health 2017, 15, 602–614.
- Stewart, V. Regulation of nitrate and nitrite reductase synthesis in enterobacteria. Antonie Van Leeuwenhoek 1994, 66, 37–45.
- Sarhadi, V.; Mathew, B.; Kokkola, A.; Karla, T.; Tikkanen, M.; Rautelin, H.; Lahti, L.; Puolakkainen, P.; Knuutila, S. Gut microbiota of patients with different subtypes of gastric cancer and gastrointestinal stromal tumors. Gut Pathog. 2021, 13, 11.
- Liu, S.; Dai, J.; Lan, X.; Fan, B.; Dong, T.; Zhang, Y.; Han, M. Intestinal bacteria are potential biomarkers and therapeutic targets for gastric cancer. Microb. Pathog. 2021, 151, 104747.
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60.
More
Information
Subjects:
Anatomy & Morphology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.3K
Entry Collection:
Gastrointestinal Disease
Revisions:
3 times
(View History)
Update Date:
21 Nov 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No