Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ravindra P Veeranna | -- | 1884 | 2022-11-11 08:27:35 | | | |
| 2 | Conner Chen | + 5 word(s) | 1889 | 2022-11-17 03:22:06 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Tulimilli, S.V.; Dallavalasa, S.; Basavaraju, C.G.; Rao, V.K.; Chikkahonnaiah, P.; Madhunapantula, S.V.; Veeranna, R.P. Structure and Genome Organization of SARS-CoV-2. Encyclopedia. Available online: https://encyclopedia.pub/entry/34926 (accessed on 08 August 2026).
Tulimilli SV, Dallavalasa S, Basavaraju CG, Rao VK, Chikkahonnaiah P, Madhunapantula SV, et al. Structure and Genome Organization of SARS-CoV-2. Encyclopedia. Available at: https://encyclopedia.pub/entry/34926. Accessed August 08, 2026.
Tulimilli, Subbarao V., Siva Dallavalasa, Chaithanya G. Basavaraju, Vinay Kumar Rao, Prashanth Chikkahonnaiah, Subbarao V. Madhunapantula, Ravindra P. Veeranna. "Structure and Genome Organization of SARS-CoV-2" Encyclopedia, https://encyclopedia.pub/entry/34926 (accessed August 08, 2026).
Tulimilli, S.V., Dallavalasa, S., Basavaraju, C.G., Rao, V.K., Chikkahonnaiah, P., Madhunapantula, S.V., & Veeranna, R.P. (2022, November 16). Structure and Genome Organization of SARS-CoV-2. In Encyclopedia. https://encyclopedia.pub/entry/34926
Tulimilli, Subbarao V., et al. "Structure and Genome Organization of SARS-CoV-2." Encyclopedia. Web. 16 November, 2022.
Copy Citation
Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2) belongs to the realm Riboviria, order Nidovirales, suborder Cornidovirineae, family Coronaviridae, subfamily Orthocoronavirinae, genus Betacoronavirus (lineage B), subgenus Sarbecovirus, and the species severe acute respiratory syndrome-related coronavirus. SARS-CoV-2 is a positive-sense, single stranded RNA virus whose genome size is ~29,903 bp.
COVID-19
SARS-CoV-2
delta
Omicron
Variants Being Monitored
1. Introduction
Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2) is a virus which surfaced in late 2019 in the city of Wuhan, China. The virus has spread across the globe quickly, leading to a pandemic situation causing acute respiratory disease known as “coronavirus disease 2019” (COVID-19) [1]. As of 21 August 2022, the ongoing pandemic has registered more than 600 million confirmed cases and 6.4 million deaths reported by the WHO [2]. The top five countries with highest numbers of cases reported are the United States of America (85,007,630), India (43,309,473), Brazil (31,611,769), France (29,164,805), and Germany (27,211,896). The top five countries with the highest number of reported deaths are the United States of America (1,002,946), Brazil (668,693), India (524,873), the Russian Federation (380,517), and Mexico (325,271) (https://covid19.who.int/ (accessed on 21 June 2022).
Although mutations are a characteristic feature of all viruses, the rate of mutations in RNA viruses is much higher than that of DNA viruses [3][4]. A study showed that the mutability magnitude of RNA viruses is five-fold compared to DNA viruses [5]. Although SARS-CoV-2, has a low sequence diversity compared to many other RNA viruses, the genetic recombination due to 3′-to-5′ exoribonuclease (nsp14) activity has produced numerous variants of SARS-CoV-2 [6][7]. In the process of evolution, a few viruses may mutate, and new variants are expected to develop over time as variants of the original virus. These variants can vary among themselves by one or more mutations [8]. A few newer variants may appear and disappear randomly and only a few may persist for a longer time [9]. The natural selection decides the destiny of the new variants [7][10]. Some mutations can negatively affect the viral replication, transmission or immunity escape, which consequently reduces the abundance of virus.
Mutations in the virus genome have an important influence on virulence, transmissibility, pathogenesis, diagnosing, treatment, as well as vaccine development [11][12]. Since “S” protein of SARS-CoV-2 is one of the primary targets in vaccine design strategy, mutations in the S region can reduce the efficacy of vaccines against the virus. Hence, mutations generated in the emerging variants should be monitored in vaccinated and non-vaccinated positive cases. Several SARS-CoV-2 variants have already been reported and documented worldwide during the COVID-19 pandemic [13]. To expand synchronization among Center for Disease Control and Prevention (CDC), National Institutes of Health (NIH), Biomedical Advanced Research Development Authority (BARDA), Food and Drug Administration (FDA) and Department of Defense (DoD), US Department of Health and Human Services (HHS) has established a SARS-CoV-2 Interagency Group (SIG). The main aim of this group is to rapidly characterize emerging variants and actively monitor their potential impact on critical SARS-CoV-2 counter-measures, including vaccines, therapeutics, and diagnostics [14]. CDC, in association with SIG has established a classification system for SARS-CoV-2 variants based on the threat level they pose to the public health and classified variants into four major types. The first type is “Variants Being Monitored (VBM)”, and the second type is “Variants of Concern (VOC)”. Variants of Interest and Variants of High Consequence (VOHC) are the third and fourth types, respectively [14]. The established nomenclature systems for naming and tracking SARS-CoV-2 lineages by GISAID, Nextstrain, and Pango are currently in use among scientific community [15]. The Nextstrain nomenclature system is based on the diversity of SARS-CoV-2 patterns and label clades that can persist for at least several months; and have a significant geographic spread [15]. The Pango lineage nomenclature is focused on the epidemiological event(s) such as the introduction of a virus into a distinct geographic area with evidence of onward spread [16]. WHO along with group of scientists (WHO Virus Evolution Working Group, the WHO COVID-19 reference laboratory network), representatives (GISAID, Nextstrain, Pango) and experts (virological, microbial nomenclature) from several countries and agencies, developed an easy-to-pronounce and non-stigmatizing labels for variants to assist discussion among public [15]. WHO expert group has recommended the use of Greek Alphabets, i.e., Alpha, Beta, Gamma, Delta (classified as previous VOC), Omicron (classified as current VOC) and Epsilon, Zeta, Eta, Theta, Lota, Kappa, Lambda and Mu (classified as previous VOI) which will be easier and more practical to be discussed by non-scientific community [15]. According to GISAID, as of 16 May 2022 187 countries shared 3,648,731 Omicron genome sequences and 203 countries shared 4,425,174 Delta genome sequences [https://www.gisaid.org/hcov19-variants/] (accessed on 16 May 2022).
The increase in the spread of VOCs has affected the efficacy of currently available vaccines. The phase III clinical trial results of the Oxford–AstraZeneca (ChAdOx1), Johnson & Johnson (Ad26.COV2.S), and Novartis (NVX-CoV2373) vaccines in South Africa have shown that the Vaccine Efficacy (VE) against the local Beta variant is decreased due to VOC [17][18][19]. For example, the protective efficacy of the Oxford–AstraZeneca (ChAdOx1) vaccine against Beta variant (mild to moderate illness) is only 10% [17]. Data from a gulf country showed that the Pfizer/BioNTech (BNT162b2) vaccine confers 87% and 72% protection against Alpha and Beta variants, respectively [20]. In Israel, with the emergence of the Delta variant, the overall efficacy of vaccines against infection has reduced from 95% to 39% [21]. In South Africa, the effectiveness of the BNT162b2 vaccine (two doses) against hospitalization for COVID-19 infection was significantly reduced from 93% in the non-Omicron period to 70% in Omicron period [22]. This decrease in the effectiveness of vaccines against the emerging VOCs has not only raised the concerns on the efficiency of vaccines but also pointed the likelihood of reinfection. Even though approved COVID-19 vaccines are less effective against the current circulating VOCs, on a brighter side, they remain to be highly effective in averting severe illness and death [23].
2. Structure and Genome Organization of SARS-CoV-2
SARS-CoV-2 virus was first reported [24] on 31 December 2019 [25]. Taxonomically, SARS-CoV-2 belongs to the realm Riboviria, order Nidovirales, suborder Cornidovirineae, family Coronaviridae, subfamily Orthocoronavirinae, genus Betacoronavirus (lineage B), subgenus Sarbecovirus, and the species severe acute respiratory syndrome-related coronavirus [26]. So far, there are seven different types of coronaviruses reported. Among these, four common human coronaviruses-229E, NL63, OC43, and HKU1—cause mild infections [27]. However, individuals infected with either of the other three coronaviruses—severe acute respiratory syndrome coronavirus (SARS-CoV), Middle East Respiratory Syndrome Coronavirus (MERSCoV), and SARS-CoV-2—develop severe respiratory distress and viral pneumonia and may ultimately succumb to the disease [28][29][30]. Jiang, Xiaowei, and Ruoqi Wang stated that although bats are the most probable reservoir animal for SARS-CoV-2 [31], zoonotic spillovers likely involving an intermediate animal and multiple transmissions from wildlife at a market in Wuhan probably led to SARS-CoV-2 emergence [32].
SARS-CoV-2 (structure shown in Figure 1A) is a positive-sense, single stranded RNA virus whose genome size is ~29,903 bp and is organized in the following order from 5’ to 3’: open reading frame (ORF) 1ab (replicas), spike glycoprotein (S), ORF3a protein, envelope protein (E), membrane glycoprotein (M), ORF6 protein, ORF7a protein, ORF7b protein, ORF8 protein, nucleocapsid-phosphoprotein (N), and ORF10 protein (Figure 1B). The SARS-CoV-2 genomic RNA contains two major open reading frames (ORFs), ORF1ab and ORF1a, occupying 2/3rd of the genome (21,291 nucleotides or 1 to 21 kb) at the 5′ end and translated to polyprotein 1ab (pp1ab) and pp1a proteins. The virus genome encodes two proteases, a papain-like protease (PLpro), or nsp3, and a 3C-like protease (3CLpro), or nsp5, which cleave pp1a and pp1b polypeptides into 16 nonstructural proteins: leader protein, nsp2, nsp3, nsp4, 3C-like proteinase, nsp6, nsp7, nsp8, nsp9, nsp10, RNA-dependent RNA polymerase (RdRp), helicase, 3’–5’ exonuclease, endoRNAse, 2’-o-ribose methyltransferase, and nsp11 [32][33]. The remaining one third of the genome (21 to 29 kb) at the 3′-end has overlapping ORFs, encoding for at least four structural proteins: spike glycoprotein (S), envelope protein (E), membrane glycoprotein (M), and nucleocapsid protein (N) and six accessory proteins (ORF3a, ORF6, ORF7a, ORF7b, ORF8, ORF10) [33][34][35]. The genes and proteins expressed by SARS-CoV-2 along with their nucleotide location and number of amino acids are shown in Table 1.
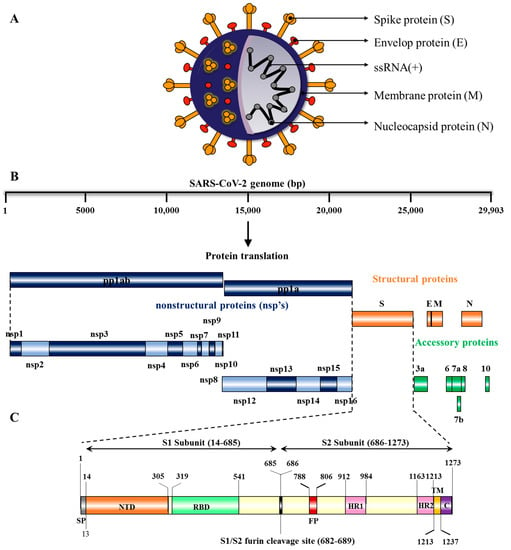
Figure 1. Genomic Organization and Structural Features of surface glycoprotein of SARS-CoV-2: (A) Structure of SARS-CoV-2: The schematic depicts the structural features of SARS-CoV-2 virus particle, which contains a single-stranded RNA, S-glycoprotein, and other structural proteins that include envelope (E), membrane (M), and nucleocapsid (N) proteins. (B) Schematic representation of SARS-CoV-2. proteins: Genome of SARS-CoV-2 consists of approximately 29,903 nucleotides, with ORF-1a and ORF-1b, which are translated to polyprotein 1a (pp1a) and 1ab (pp1ab), respectively. Four genes encoding for structural proteins such as spike (S), envelop (E), membrane (M), and nucleocapsid (N). Accessory proteins (ORF3a, 6, 7a, 7b, 8, and 10) are distributed among structural proteins. (C) Structure of SARS-CoV-2 Surface glycoprotein (S): The surface glycoprotein (S protein) is made up of 1273 amino acids, including N-terminal signal peptide (SP), S1 subunit, and S2 subunit. The S1 subunit contains an N-terminal domain (NTD) and a receptor binding domain (RBD), while the S2 subunit is composed of the fusion peptide (FP), heptapeptide repeat sequence 1 (HR1), HR2, TM domain, and cytoplasm domain (C).
Table 1. Genes and proteins expressed by SARS-CoV-2.
| Number | Gene | Nucleotide Location | Protein | Amino Acids |
|---|---|---|---|---|
| 1 | ORF1a | 266–13,483 | Polyprotein 1a | 4405 |
| 1 | ORF1ab | 266–21,555 | Polyprotein 1ab | 7096 |
| 2 | ORF2 | 21,563–25,384 | Surface glycoprotein or Spike (S protein) |
1273 |
| 3 | ORF3a | 25,393–26,220 | ORF3a protein | 275 |
| 4 | ORF4 | 26,245–26,472 | Envelope protein (E protein) | 75 |
| 5 | ORF5 | 26,523–27,191 | Membrane glycoprotein (M protein) |
222 |
| 6 | ORF6 | 27,202–27,387 | ORF 6 protein | 61 |
| 7 | ORF7a | 27,394–27,759 | ORF 7a protein | 121 |
| 8 | ORF7b | 27,756–27,887 | ORF 7b protein | 43 |
| 9 | ORF8 | 27,894–28,259 | ORF 8 protein | 121 |
| 10 | ORF9 | 28,274–29,533 | Nucleocapsid phosphoprotein (N protein) |
419 |
| 11 | ORF10 | 29,558–29,674 | ORF10 protein | 38 |
The surface glycoprotein (S protein) is composed of 1273 amino acids, including the N-terminal signal peptide (SP) of 1–13 residues, the S1 subunit (14–685 residues) and S2 subunit (686–1273 residues) (Figure 1C). The S1 subunit is made up of an N-terminal domain (NTD, 14–305 residues) and a receptor binding domain (319–541 residues), while the S2 subunit is made up of the fusion peptide (FP, 788–806 residues), heptapeptide repeat sequence 1 (HR1, 912–984 residues), HR2 (1163–1213 residues), TM domain (1213–1237 residues) and cytoplasm domain (1237–1273 residues) [36]. Both S1 and S2 subunits are crucial in assembly and surface projection of the S protein, which interacts with Angiotensin-Converting Enzyme 2 (ACE2) receptors which are expressed on the lower respiratory pneumocytes of the host cell [30][33]. Host transmembrane Serine Protease 2 (TMPRSS2) cleaves the S protein at the furin cleavage site (682–689 residues) into S1 and S2 subunits, enabling viral fusion and entry [37][38] (Figure 2). After entering into the host cell, SARS-CoV-2 takeovers the host cell machinery to rapidly synthesize viral proteins, assemble, and release virus progenies [39][40].
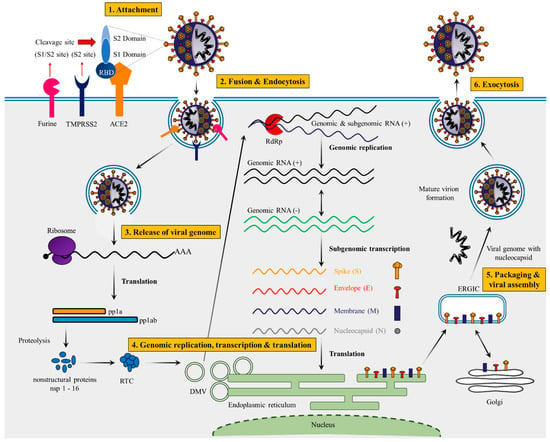
Figure 2. Complete life cycle of SARS-CoV-2. SARS-CoV-2 lifecycle begins with primary binding of the Spike protein to its specific receptors (ACE2). Mostly host cell entry depends on several steps: (i) Initial cleavage of S1/S2 specific sites by surface transmembrane protease serine-2 (TMPRSS2) and furine, (ii) followed by virus–cell membrane fusion and endocytosis. (iii) Continuing from endocytosis, m-RNA genome is mainly released into the cytosol and translated into the polyproteins. Mostly polyproteins (pp1a and pp1ab) are cleaved by specific viral-encoded protease (VEP) into the several nonstructural proteins (nsps) (mostly including RNA-dependent RNA polymerase: RdRp) which is responsible for replication transcription complex (RTC) in the cells. (iv) Viral replication begins with virus-induced double membrane vesicles mainly derived from the endoplasmic reticulum (ER). (v) Positive-strand of genome serves as a main template for full-length negative-strand RNA and sub genomic (sg)RNA and sgRNA translation results in both structural proteins and accessory proteins are inserted into the ER–Golgi intermediate compartment (ERGIC) for virion assembly, respectively. (vi) RNA genomes with specific nucleocapsid proteins are incorporated into newly synthesized virions, which are secreted by exocytosis. DMV—double-membrane vesicle.
References
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021, 19, 141–154.
- World Health Organization. WHO Coronavirus Disease (COVID-19) Dashboard. Available online: https://covid19.who.int/ (accessed on 29 August 2021).
- Lauring, A.S.; Hodcroft, E.B. Genetic Variants of SARS-CoV-2-What Do They Mean? JAMA 2021, 325, 529–531.
- Almubaid, Z.; Al-Mubaid, H. Analysis and comparison of genetic variants and mutations of the novel coronavirus SARS-CoV-2. Gene Rep. 2021, 23, 101064.
- Duffy, S.; Shackelton, L.A.; Holmes, E.C. Rates of evolutionary change in viruses: Patterns and determinants. Nat. Rev. Genet. 2008, 9, 267–276.
- Gribble, J.; Stevens, L.J.; Agostini, M.L.; Anderson-Daniels, J.; Chappell, J.D.; Lu, X.; Pruijssers, A.J.; Routh, A.L.; Denison, M.R. The coronavirus proofreading exoribonuclease mediates extensive viral recombination. PLoS Pathog. 2021, 17, e1009226.
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827.e819.
- World Health Organization. An Update on SARS-CoV-2 Virus Mutations & Variants. Available online: https://www.who.int/docs/default-source/coronaviruse/risk-comms-updates/update47-sars-cov-2-variants.pdf?sfvrsn=f2180835_4 (accessed on 25 March 2021).
- Centers for Disease Control and Prevention. About Variants of the Virus that Causes COVID-19. Available online: https://www.cdc.gov/coronavirus/2019-ncov/transmission/variant.html (accessed on 25 March 2021).
- Su, S.; Wong, G.; Shi, W.; Liu, J.; Lai, A.C.K.; Zhou, J.; Liu, W.; Bi, Y.; Gao, G.F. Epidemiology, Genetic Recombination, and Pathogenesis of Coronaviruses. Trends Microbiol. 2016, 24, 490–502.
- Chakraborty, S. Evolutionary and structural analysis elucidates mutations on SARS-CoV2 spike protein with altered human ACE2 binding affinity. Biochem. Biophys. Res. Commun. 2021, 534, 374–380.
- Hou, Y.J.; Chiba, S.; Halfmann, P.; Ehre, C.; Kuroda, M.; Dinnon, K.H., 3rd; Leist, S.R.; Schafer, A.; Nakajima, N.; Takahashi, K.; et al. SARS-CoV-2 D614G variant exhibits efficient replication ex vivo and transmission in vivo. Science 2020, 370, 1464–1468.
- Burki, T. Understanding variants of SARS-CoV-2. Lancet 2021, 397, 462.
- Centers for Disease Control and Prevention. SARS-CoV-2 Variant Classifications and Definitions. Available online: https://www.cdc.gov/coronavirus/2019-ncov/variants/variant-classifications.html?CDC_AA_refVal=https%3A%2F%2Fwww.cdc.gov%2Fcoronavirus%2F2019-ncov%2Fvariants%2Fvariant-info.html (accessed on 25 August 2021).
- World Health Organization. Tracking SARS-CoV-2 variants. Available online: https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/ (accessed on 25 August 2021).
- Rambaut, A.; Holmes, E.C.; O’Toole, A.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407.
- Madhi, S.A.; Baillie, V.; Cutland, C.L.; Voysey, M.; Koen, A.L.; Fairlie, L.; Padayachee, S.D.; Dheda, K.; Barnabas, S.L.; Bhorat, Q.E.; et al. Efficacy of the ChAdOx1 nCoV-19 Covid-19 Vaccine against the B.1.351 Variant. N. Eng. J. Med. 2021, 384, 1885–1898.
- Shinde, V.; Bhikha, S.; Hoosain, Z.; Archary, M.; Bhorat, Q.; Fairlie, L.; Lalloo, U.; Masilela, M.S.; Moodley, D.; Hanley, S. Efficacy of NVX-CoV2373 Covid-19 vaccine against the B. 1.351 variant. N. Eng. J. Med. 2021, 384, 1899–1909.
- Sadoff, J.; Gray, G.; Vandebosch, A.; Cárdenas, V.; Shukarev, G.; Grinsztejn, B.; Goepfert, P.A.; Truyers, C.; Fennema, H.; Spiessens, B. Safety and efficacy of single-dose Ad26. COV2. S vaccine against Covid-19. N. Eng. J. Med. 2021, 384, 2187–2201.
- Abu-Raddad, L.J.; Chemaitelly, H.; Butt, A.A.; National Study Group for COVID-19 Vaccination. Effectiveness of the BNT162b2 Covid-19 Vaccine against the B.1.1.7 and B.1.351 Variants. N. Eng. J. Med. 2021, 385, 187–189.
- Haas, E.J.; Angulo, F.J.; McLaughlin, J.M.; Anis, E.; Singer, S.R.; Khan, F.; Brooks, N.; Smaja, M.; Mircus, G.; Pan, K.; et al. Impact and effectiveness of mRNA BNT162b2 vaccine against SARS-CoV-2 infections and COVID-19 cases, hospitalisations, and deaths following a nationwide vaccination campaign in Israel: An observational study using national surveillance data. Lancet 2021, 397, 1819–1829.
- Collie, S.; Champion, J.; Moultrie, H.; Bekker, L.-G.; Gray, G. Effectiveness of BNT162b2 vaccine against omicron variant in South Africa. N. Eng. J. Med. 2022, 386, 494–496.
- Lauring, A.S.; Tenforde, M.W.; Chappell, J.D.; Gaglani, M.; Ginde, A.A.; McNeal, T.; Ghamande, S.; Douin, D.J.; Talbot, H.K.; Casey, J.D.; et al. Clinical Severity and mRNA Vaccine Effectiveness for Omicron, Delta, and Alpha SARS-CoV-2 Variants in the United States: A Prospective Observational Study. medRxiv 2022.
- Li, X.; Zai, J.; Wang, X.; Li, Y. Potential of large "first generation" human-to-human transmission of 2019-nCoV. J. Med. Virol. 2020, 92, 448–454.
- Gralinski, L.E.; Menachery, V.D. Return of the Coronavirus: 2019-nCoV. Viruses 2020, 12, 135.
- Chan, J.F.; Kok, K.H.; Zhu, Z.; Chu, H.; To, K.K.; Yuan, S.; Yuen, K.Y. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect. 2020, 9, 221–236.
- Fehr, A.R.; Perlman, S. Coronaviruses: An overview of their replication and pathogenesis. Methods Mol. Biol. 2015, 1282, 1–23.
- Tay, M.Z.; Poh, C.M.; Renia, L.; MacAry, P.A.; Ng, L.F.P. The trinity of COVID-19: Immunity, inflammation and intervention. Nat. Rev. Immunol. 2020, 20, 363–374.
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. Author Correction: A new coronavirus associated with human respiratory disease in China. Nature 2020, 580, E7.
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273.
- Temmam, S.; Montagutelli, X.; Hérate, C.; Donati, F.; Regnault, B.; Attia, M.; Salazar, E.B.; Chrétien, D.; Conquet, L.; Jouvion, G. SARS-CoV-2-Related Bat Virus in Human Relevant Models Sheds Light on the Proximal Origin of COVID-19. Res. Sq. 2022. preprint. Available online: https://assets.researchsquare.com/files/rs-1803095/v1_covered.pdf?c=1657179682 (accessed on 1 July 2022).
- Jiang, X.; Wang, R. Wildlife trade is likely the source of SARS-CoV-2. Science 2022, 377, 925–926.
- Wu, A.; Peng, Y.; Huang, B.; Ding, X.; Wang, X.; Niu, P.; Meng, J.; Zhu, Z.; Zhang, Z.; Wang, J.; et al. Genome Composition and Divergence of the Novel Coronavirus (2019-nCoV) Originating in China. Cell Host Microbe 2020, 27, 325–328.
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468.
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224.
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.
- Matsuyama, S.; Nao, N.; Shirato, K.; Kawase, M.; Saito, S.; Takayama, I.; Nagata, N.; Sekizuka, T.; Katoh, H.; Kato, F.; et al. Enhanced isolation of SARS-CoV-2 by TMPRSS2-expressing cells. Proc. Natl. Acad. Sci. USA 2020, 117, 7001–7003.
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.C.; Uhl, S.; Hoagland, D.; Moller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, 1036–1045.
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448.
- Guo, Y.R.; Cao, Q.D.; Hong, Z.S.; Tan, Y.Y.; Chen, S.D.; Jin, H.J.; Tan, K.S.; Wang, D.Y.; Yan, Y. The origin, transmission and clinical therapies on coronavirus disease 2019 (COVID-19) outbreak - an update on the status. Mil. Med. Res. 2020, 7, 11.
More
Information
Subjects:
Infectious Diseases
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.0K
Entry Collection:
COVID-19
Revisions:
2 times
(View History)
Update Date:
17 Nov 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No