Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Lilla Gombos | -- | 2677 | 2022-11-16 10:51:19 | | | |
| 2 | Rita Xu | Meta information modification | 2677 | 2022-11-17 02:35:40 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Gombos, L.G.; Waldvogel, S.R. Electrochemical Bromofunctionalization of Alkenes. Encyclopedia. Available online: https://encyclopedia.pub/entry/34913 (accessed on 09 August 2026).
Gombos LG, Waldvogel SR. Electrochemical Bromofunctionalization of Alkenes. Encyclopedia. Available at: https://encyclopedia.pub/entry/34913. Accessed August 09, 2026.
Gombos, Lilla G., Siegfried R. Waldvogel. "Electrochemical Bromofunctionalization of Alkenes" Encyclopedia, https://encyclopedia.pub/entry/34913 (accessed August 09, 2026).
Gombos, L.G., & Waldvogel, S.R. (2022, November 16). Electrochemical Bromofunctionalization of Alkenes. In Encyclopedia. https://encyclopedia.pub/entry/34913
Gombos, Lilla G. and Siegfried R. Waldvogel. "Electrochemical Bromofunctionalization of Alkenes." Encyclopedia. Web. 16 November, 2022.
Copy Citation
The electrochemical generation of highly reactive and hazardous bromine under controlled conditions as well as the reduction of surplus oxidizers and reagent waste has placed electrochemical synthesis in a highlighted position. In particular, the electrochemical dibromination and bromofunctionalization of alkenes and alkynes have received significant attention, as the forming of synthetically important derivatives can be generated from bench-stable and safe bromide sources under “green” conditions.
anode
alkenes
bromine
electrochemistry
1. Introduction
With the increasing amount of economic, environmental and political pressure, the chemical industry faces the challenge of providing the public and the scientific community a sustainable and greener alternative to conventional chemical methods [1]. Electro-organic synthesis has become one of the most attractive research topics in recent years, providing highly versatile synthetic methodologies [2][3][4][5][6]. Using electrons as “traceless” redox reagents allows the elimination of the excess use of harmful redox chemicals with a high atom efficiency [7]. The electro-generation of highly reactive species in-situ allows the utilization of inherently safe surrogates and provides a simplified approach [8]. These advantages, in combination with the valorization of renewable feedstock and “green electricity”, makes electrochemical functionalization particularly attractive.
2. Electrochemical Bromofunctionalization of Alkenes
2.1. HBr, MBr and Alkylammonium Bromides as Bromine Sources
Pioneering work in the field was demonstrated by Torii in the early 80s, reporting the bromofunctionalization of alkenes in the presence of aq. NaBr, which popularized the indirect electrochemical oxidation of olefins using stable and inexpensive metal halide salts [9][10][11][12][13]. This form of a bromine source has received particular attention due to its ability to tame hazardous and toxic molecular bromine in-situ under controlled conditions. It is worth mentioning that the complementary half-reaction´s electrode choice is usually a low-hydrogen-overpotential material such as platinum or nickel, which supports cathodic hydrogen evolution. Nevertheless, platinum group metals are depleting, costly and highly contaminating [14][15][16].
In 2019 Hu, Fang and Mei reported the electrochemical radical formyloxylation–bromination of various alkenes in a regio- and chemoselective fashion [17]. The implementation of stable NaBr as a radical bromine source makes this approach sustainable. DMF serves a dual role as both the solvent and the formyloxylation reagent, while acetic acid supports the cathodic reaction. Para-substituted electron-rich styrene derivatives afforded excellent yields, while electron-deficient and strongly electron-withdrawing groups prolonged the reaction time. The naturally derived safrole-type and estrone-type derivates also readily underwent electrochemical formyloxylation, proving the possibility for the late-stage functionalization of pharmaceutical scaffolds. In total, 30 examples were reported with excellent yields up to 97%. The scalability was proved via a gram-scale experiment (6.0 mmol) resulting in an isolated yield of 90%. The method was extended to the formyloxylation–chlorination and formyloxylation–trifluoromethylation of alkenes as well. The authors suggest that bromide is oxidized at the anode to bromo-radicals, which then rapidly combine with arylalkene 1 to form the benzyl radical I. Via a second oxidation, the forming benzylic carbocation II is subjected to nucleophilic attack by DMF to iminium intermediate III, which is subsequently hydrolyzed resulting in the formyloxylated brominated product 2 (Scheme 1).
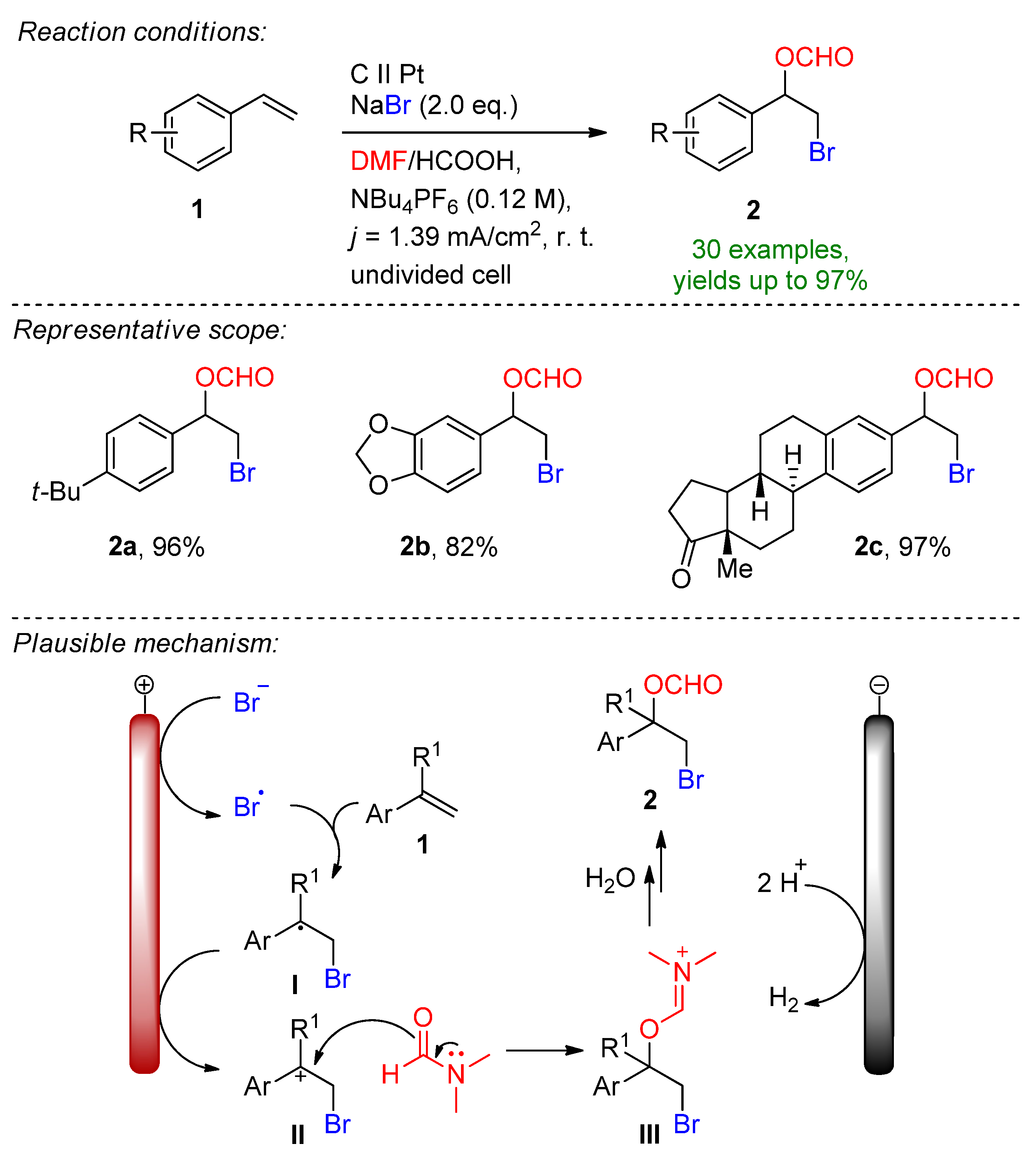
Scheme 1. Electrochemical radical formyloxylation bromination of alkenes [17].
The electrobiocatalytic bromolactonization was reported by Bormann and Holtmann on carbon-nanotube-modified gas-diffusion electrodes [18]. The paper reports the cathodic reduction of ambient oxygen to H2O2 at a reduced overpotential in a divided cell separated via a proton exchange membrane. Curvularia inaequalis (CiVCPO), a vanadium-dependent chloroperoxidase enzyme converts the forming peroxide in the presence of potassium bromide into hypobromite, which leads to the bromolactonization of 5-pentenoic acid. The reaction is complimented via the oxidation of water at a platinum anode. The superiority of the reaction lies in the oxygen-enriched, modified-carbon-nanotube diffusion electrode that significantly lowers the overpotential for peroxide formation, which is a sensible factor for the enzymatic catalytic reactivity [19].
The electrochemical bromocyclization of tryptophol, tryptamine and tryptophan derivatives was recently reported by Wu and Vincent [20]. This efficient protocol features MgBr2 with a dual role as a halogen source and supporting electrolyte. The reaction proceeds in an undivided cell under both constant current (CCE) and constant potential (CPE) conditions in the presence of 1 eq. MgBr2. Magnesium bromide is oxidized at the carbon anode into bromine in an acetonitrile/water solution under ambient conditions. It is worth mentioning that under CPE, the constant potential window is maintained via measuring the potential against a reference electrode. Here, the constant terminal voltage of the cell was determined via measuring the constant terminal voltage between the electrodes, which is not an adequate parameter for CPE conditions. The dearomative reaction proceeds via the formation of bromonium intermediate IV, followed by intramolecular cyclization to form the corresponding brominated derivatives with excellent yields (Scheme 2). This simple and environmentally friendly set up featured 22 examples with up to 96% isolated yields and a broad scope of functional group tolerance.
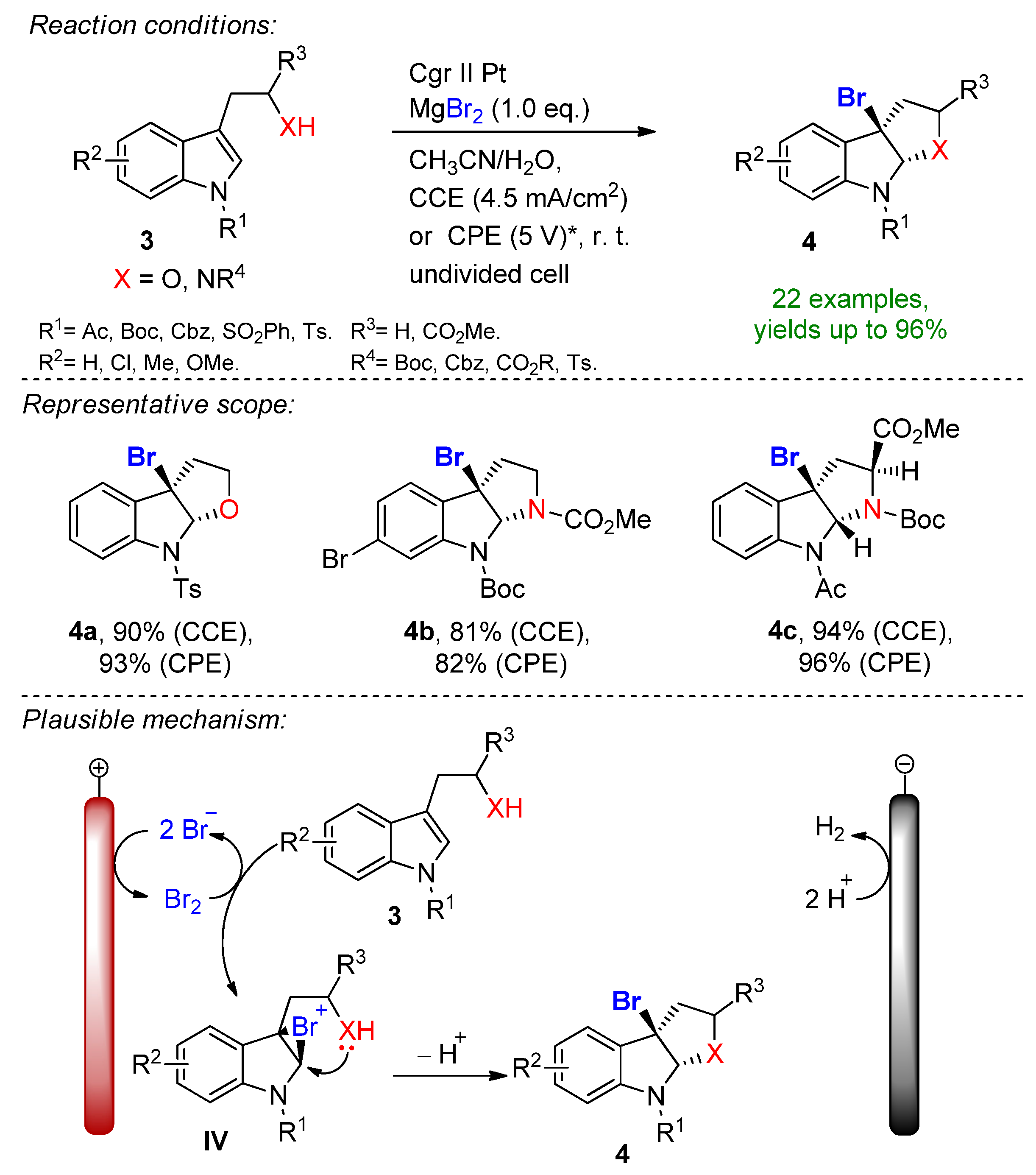
Scheme 2. The electrochemical bromocyclization of tryptophol, tryptamine and tryptophan derivates utilizing MgBr2. * Constant terminal voltage of the cell was observed between a graphite anode and platinum plate cathode [20]. Cgr = graphite.
The synthetic utility was demonstrated via the electrochemical bromocyclization and further functionalization of L-tryptophan-derived diketopiperazine (5) to (−)-epi-amauromine (7a) and (+)-novoamauromine (7b), which was reported to be superior to the conventional procedure using stoichiometric NBS (Scheme 3) [21].
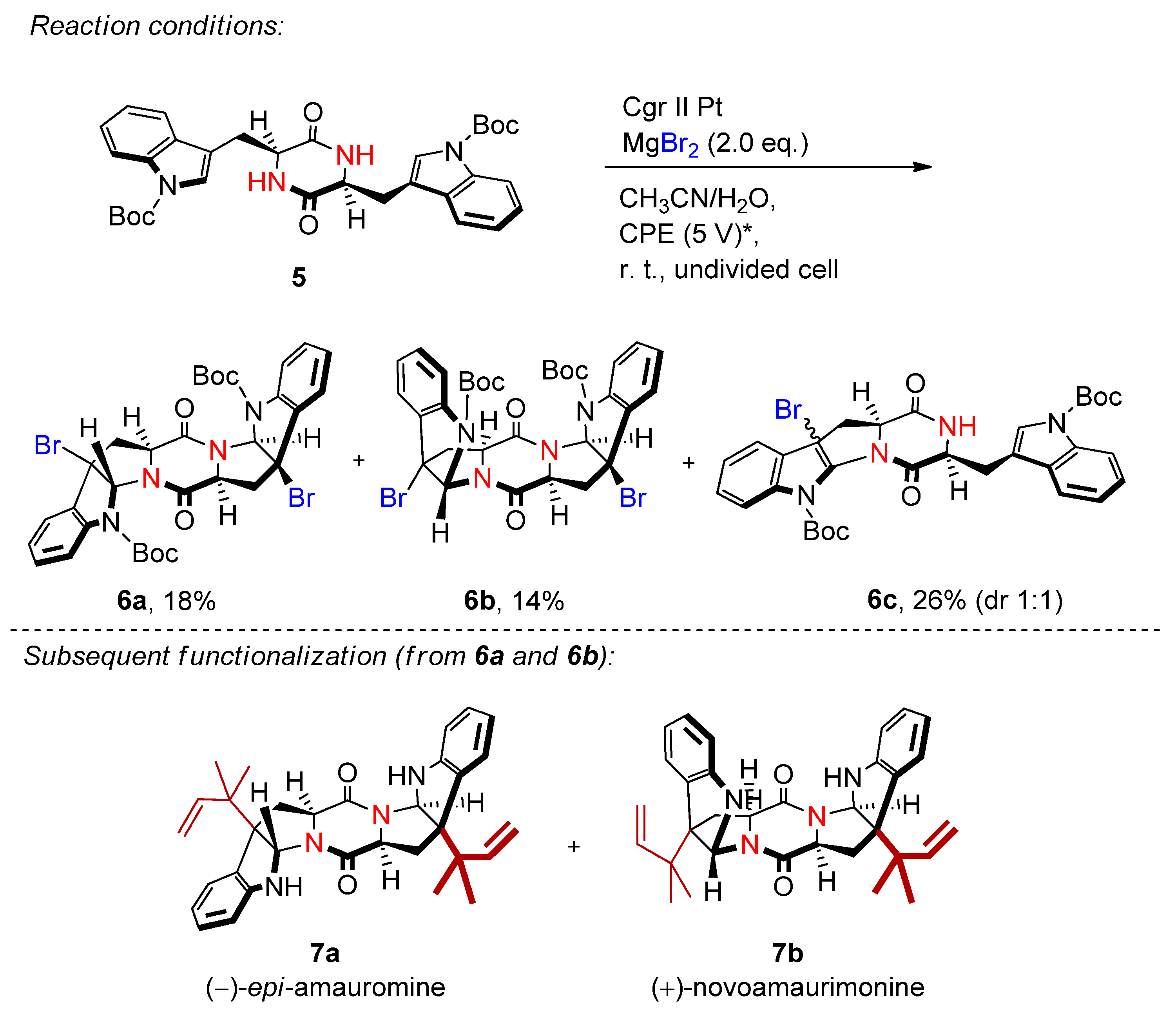
Solvent stabilizing effects have been reported in electrochemical synthesis, for example using hexafluoroisopropanol (HFIP) [22][23][24][25][26][27]. Yoshida demonstrated the formation of α-bromocarbonyls and bromohydrins via a low-temperature DMSO-stabilized halogen cation pool method [28][29]. Based on these reports, a sophisticated technique for the electrochemical bromohydrin and bromohydrin ether formation was reported in a chemoselective fashion (Scheme 4) [30]. The anodic oxidation of potassium bromide on a glassy carbon electrode in the presence of 10 eq. acetic acid in DMSO/H2O allowed the corresponding bromohydrins to be produced in excellent yields. By changing the solvent system to acetonitrile/alcohol, bromohydrin ethers were obtained. The key additive of this approach is the addition of acetic acid or trifluoroacetic acid. In the absence of the additive, the yield drops dramatically. The reaction is complemented with hydrogen evolution at a platinum cathode.
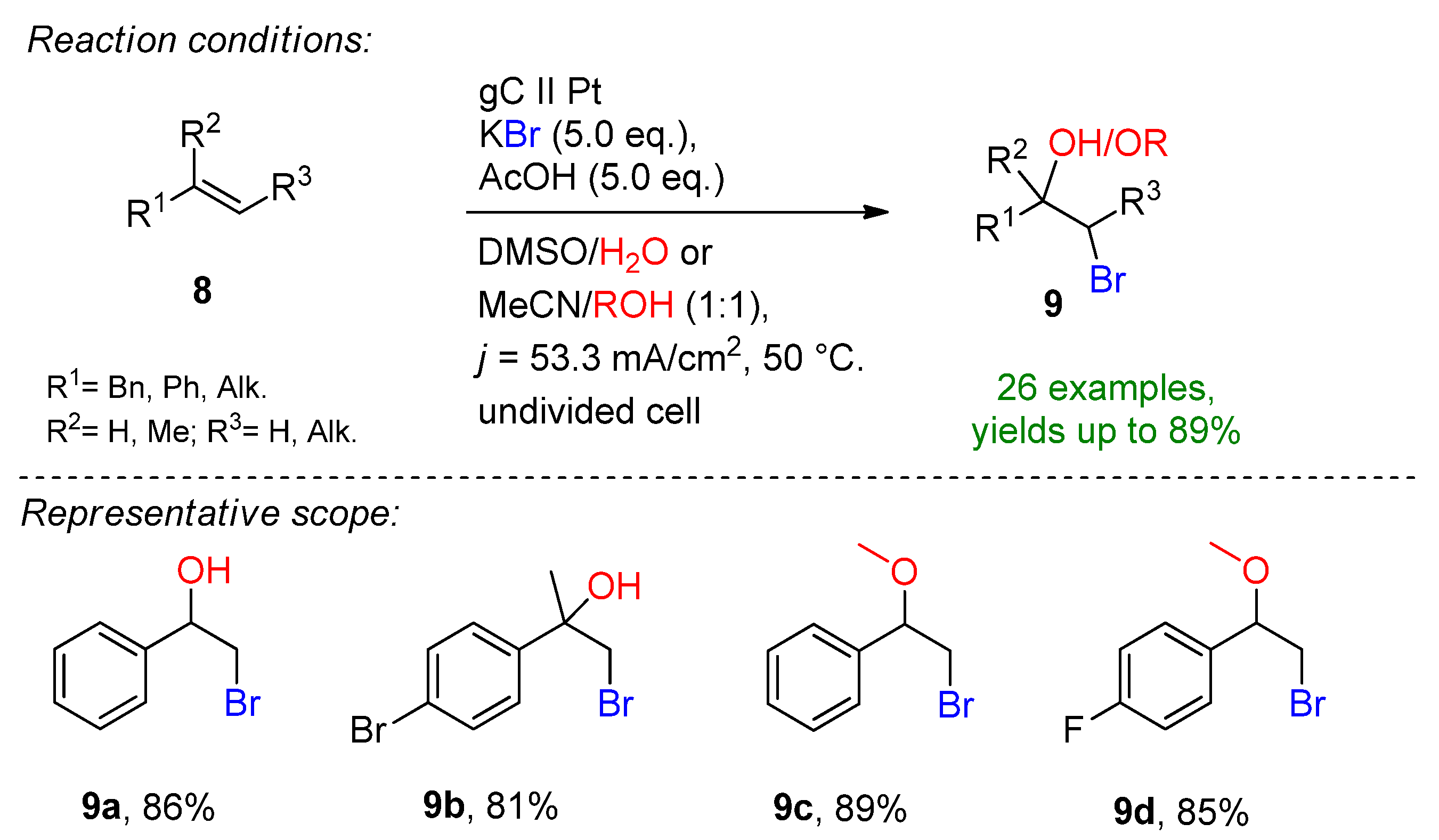
Scheme 4. Electrochemical bromohydrin and bromohydrin ether formation [30]. gC = glassy carbon.
Sun and co-workers reported an elegant way of on-site bromination and hydrogenation in a simultaneous fashion [31]. The paired electrolysis takes place in a H-type cell setup divided by a Nafion membrane. This method demonstrates high halogenation flexibility and functional group tolerance, as the bromination reaction is spatially separated from the electrolysis event. The key principle is the use of bench-stable NaBr as a halogen source and the utilization of cathodic hydrogen evolution to achieve a high atom economy and energy efficiency. The anodically generated bromine gas from aqueous sodium bromide in acidic media is transferred into a separate compartment, where ideal conditions for the corresponding substrates can be set up. Likewise, cathodically generated hydrogen gas is used in the presence of Pt/C, demonstrating the synergic pairing of on-site bromination and hydrogenation. This is an excellent example for the utilization of the redox properties of the electrochemical cell.
Similarly, Hilt and his group published a linear paired electrolysis for the electrochemical dibromination of alkenes with the realization of 200% current efficiency for stoichiometric transformations in the presence of oxygen [32]. The dual role supporting electrolyte and bromide source NBu4Br is used as a brominating agent. Bromide undergoes direct oxidation at the anode to bromine and is produced mediated via reductively formed H2O2 determining the theoretical applied charge of 1 F (Scheme 5). The stable tribromide species are the results of the stabilizing effect of acetonitrile and the high concentration of bromide. Using this method, the authors reported 13 examples with good to excellent yields, providing 11c quantitatively with a current efficiency of 200%. The reaction proved to have a good functional group tolerance via performing the Glorius-test [33][34]. The method could be adopted to the bromination of arenes and for the iodination of alkenes as well.
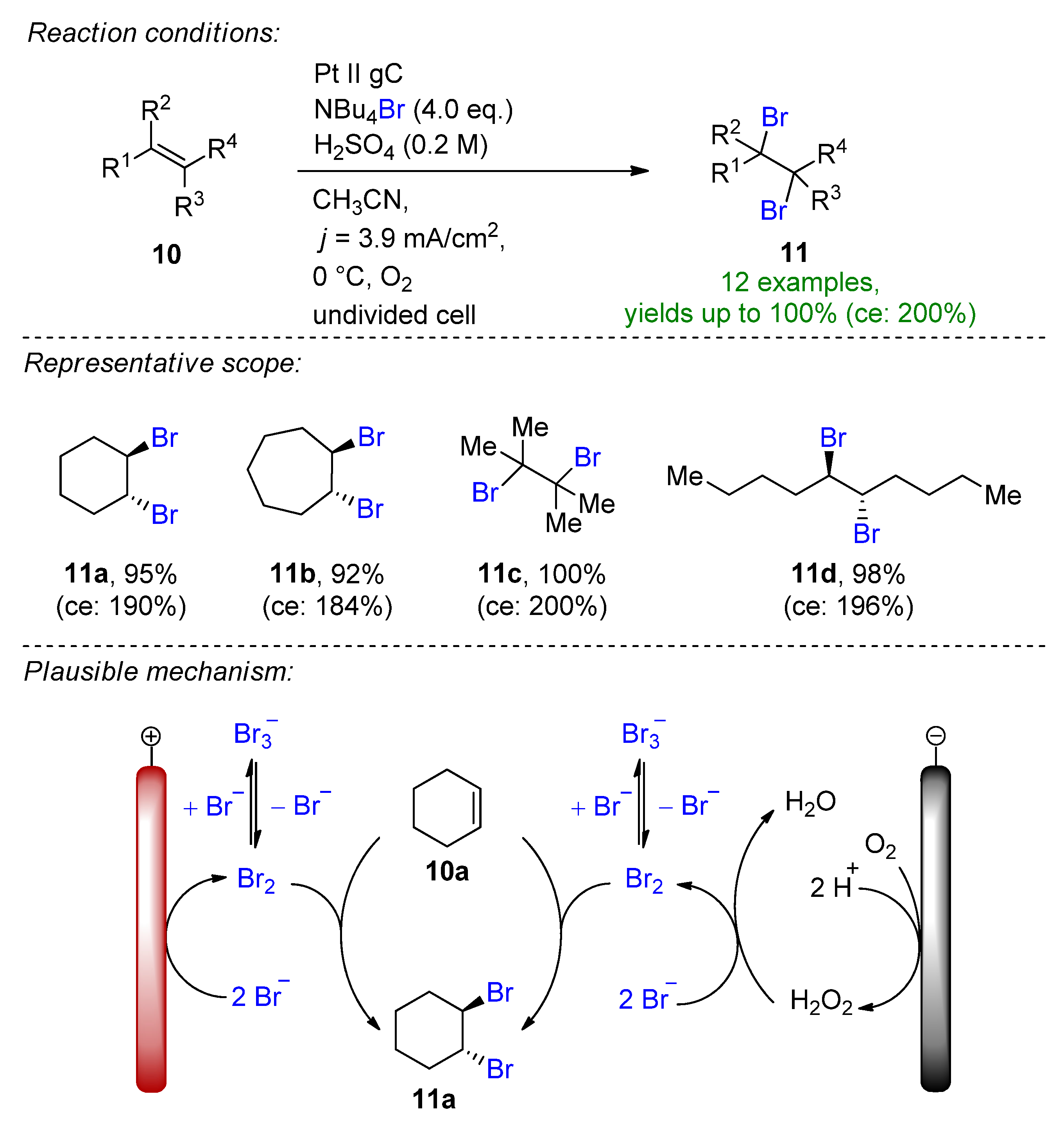
Scheme 5. Linear paired electrolysis for the dibromination of alkenes [32]. gC = glassy carbon.
A highly regioselective electrochemical protocol for the synthesis of isoxazolines from β,γ-unsaturated ketoximes via cascade C–O and C–Br annulation was reported (Scheme 6) [35]. The stable inorganic potassium bromide has a dual role of serving as a bromine source as well as a supporting electrolyte. The key features of this method are the generation of DMSO-stabilized bromine in-situ and the employment of a sodium acetate base under high current density conditions to form the desired bromoethyl-substituted isoxazolines. The method demonstrated excellent functional group tolerance and a high selectivity, and the isoxazoline derivatives could be obtained with up to 81% isolated yields. The presence of the base as well as the electrode material choice is crucial to the success of the reaction. The scalability in both batch-type and flow electrolysis were demonstrated.
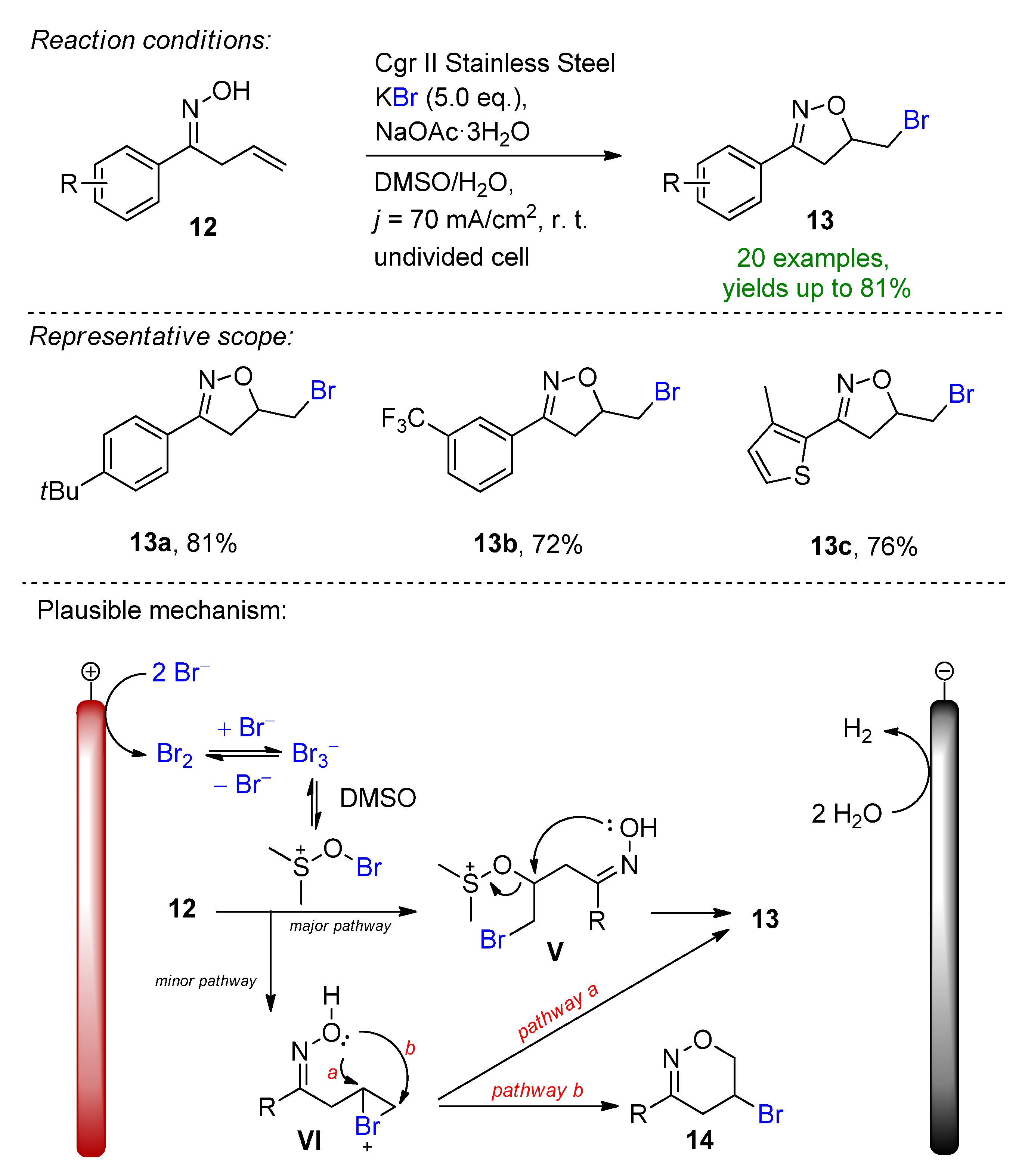
Scheme 6. Electrochemical formation of isoxazolines with the aid of KBr [35]. Cgr = graphite.
Yin and co-workers introduced an oxidant- and base-free electrochemical intramolecular halo-amination of unactivated alkenes to form diverse brominated N-heterocycles [36]. They provide a simple electrochemical protocol using bench-stable LiBr or LiI as a halide source and a supporting electrolyte furnishing a dual role. The reaction proceeded smoothly at room temperature, providing 26 examples combined with the iodo-cyclization in excellent yields. The reaction tolerates labile functional groups such as cyclopropyl, substituted aromatics and heterocycles. Moreover, in the presence of LiI, highly challenging N-heterocycles such as the three-membered aziridine or six-membered piperidine could be formed (Scheme 7).
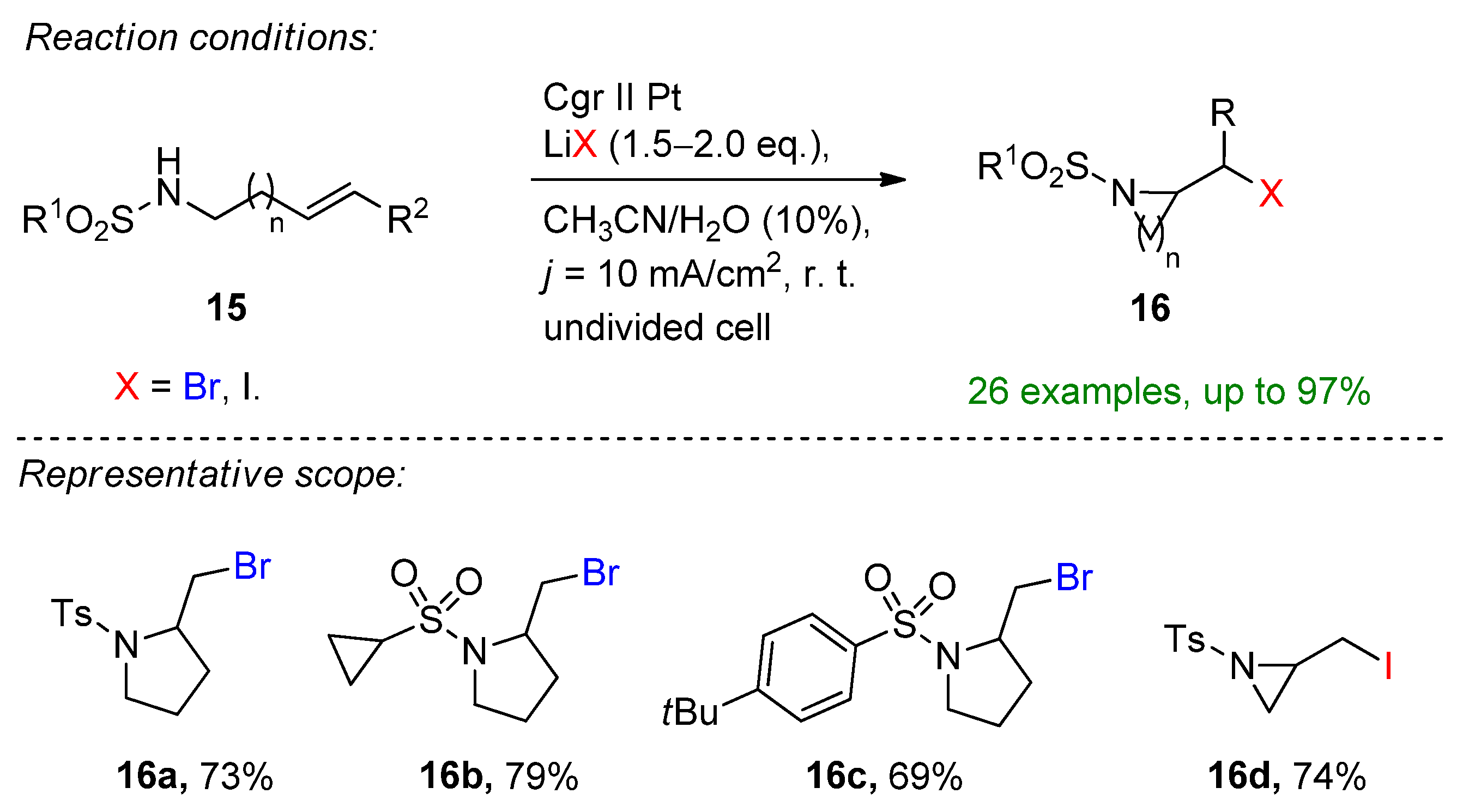
Scheme 7. Electrochemical intramolecular haloamination with the aid of LiBr and LiI [36]. Cgr = graphite.
Similarly, the electrochemical oxidative bromolactonization of unsaturated carboxylic acids was reported for the first time [37]. This environmentally friendly approach features sodium bromide as the halogen source in acetonitrile, which is oxidized at the carbon rod anode to give the corresponding bromine radical, which directly reacts with the alkene of the carboxylic acid to form intermediate V (Scheme 8). After subsequent oxidation to the stabilized cationic intermediate VI and cyclization, the corresponding bromoethylated γ-lactones could be formed with excellent yields. Control experiments also supported the proposed mechanism, as in the presence of radical scavengers or molecular bromine, the yield dropped dramatically. The authors propose hydrogen evolution as a cathodic counter-reaction at a nickel electrode.
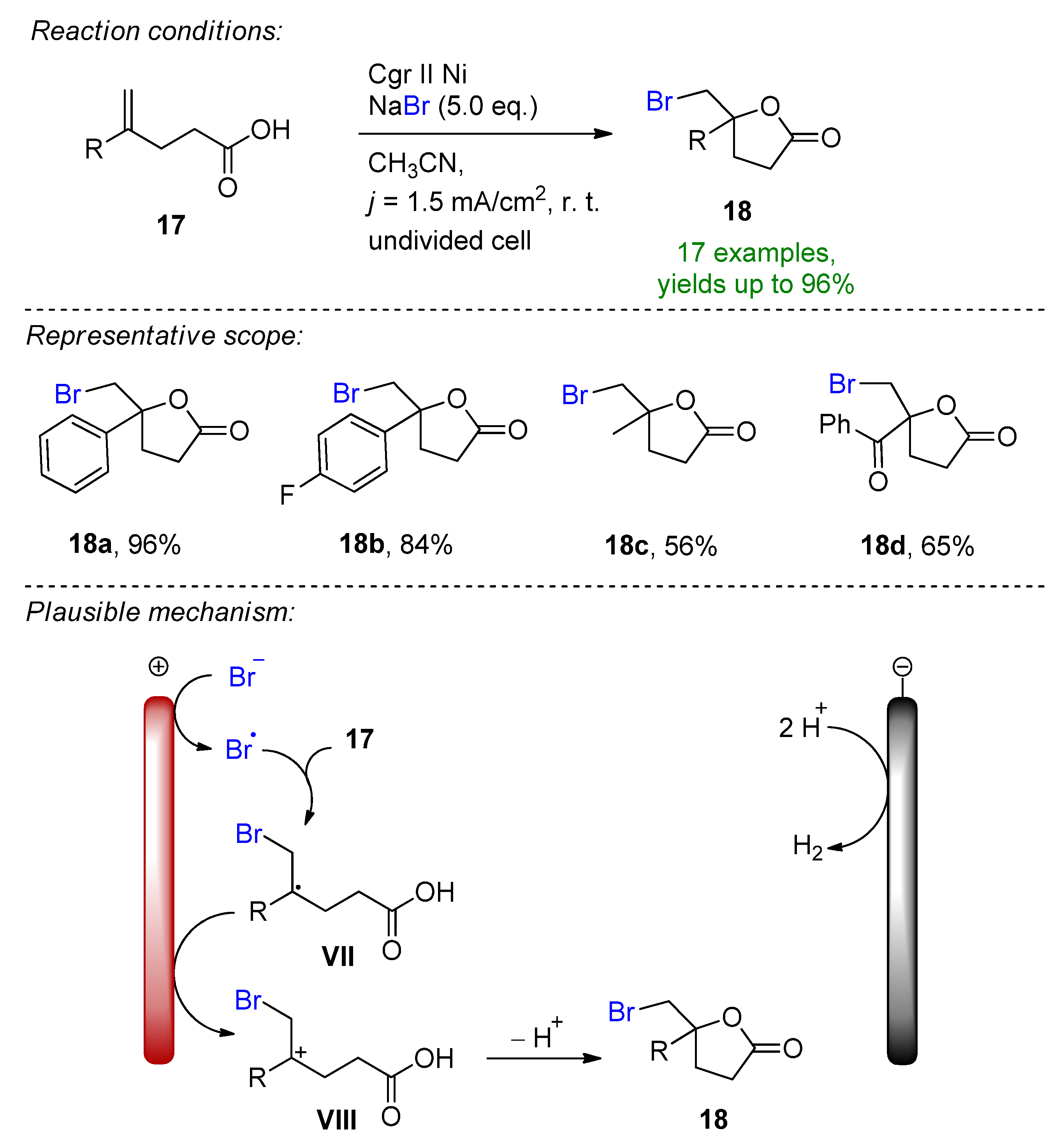
Scheme 8. Electrochemical bromolactonization of unsaturated carboxylic acids [37]. Cgr = graphite.
The electrochemical bromination of electron-deficient alkenes in quinones, coumarins, quinoxalines and 1,3-diketones has also been reported [38]. The synthetically useful organohalides could be obtained using bromides on graphite felt in combination with a platinum cathode. The key feature of this bromination method is the in-situ formation of HBr from KBr and H2SO4, which is anodically oxidized to bromine. Control experiments with 2,6-di-tert-butyl-4-methylphenol suggest a radical pathway by the formation of the halogen.
Recently, Waldvogel et al. published the selective electrochemical bromination of terpenes and naturally derived alkenes [39]. The challenging halofunctionalization of renewable feedstock was demonstrated by the employment of inexpensive and bench-stable NaBr with a dual role of being both a bromine source and supporting electrolyte. The test substrate, limonene, could be brominated selectively under ambient conditions giving the brominated derivative in a 53% yield. The optimization procedure showed that the careful consideration of the MeCN/H2O solvent system as well as the NaBr as a bromide source are important features of the method. Other inorganic or organic bromide salts resulted in a diminished yield or no conversion. The method could be successfully extended to linear and cyclic monoterpenes, terpenoids and phenylpropanoids to give 10 desired vicinal 1,2-dibromo derivatives, with the bromination of carvone representing the best yield of 82% (20b, Scheme 9). A slight change in the electrochemical parameters provided the tetrabrominated limonene derivative in a 74% isolated yield. The reaction proved to be scalable and a synthetic utility could be demonstrated via subsequent functionalization to the α,β-unsaturated nitrile derivative 21a for the first time. Cyclic voltammetry studies support the oxidative bromine formation in-situ, which is complimented by the liberation of H2 at the cathode.
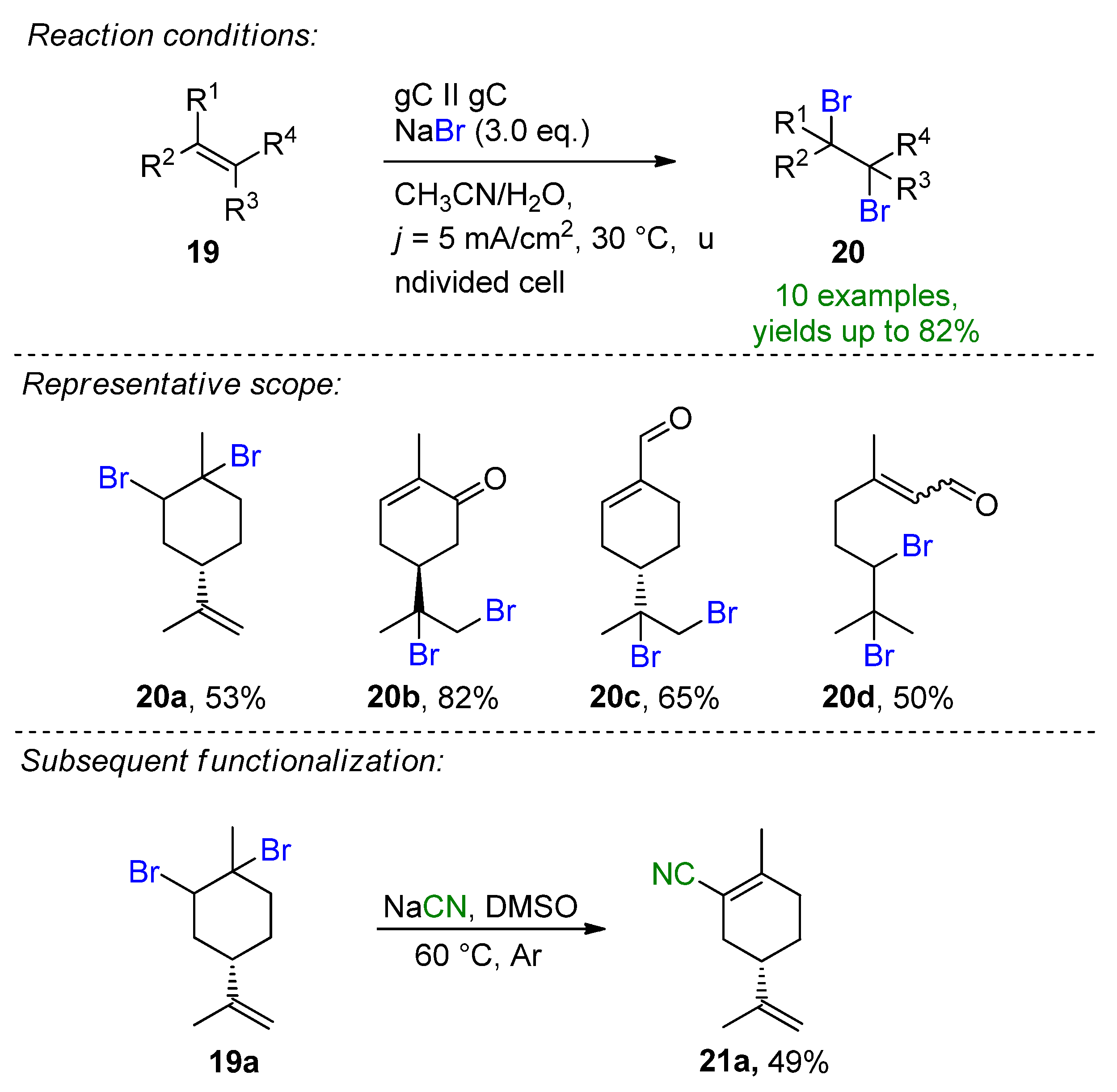
Scheme 9. Electrochemical dibromination of terpenes and terpenoids [39]. gC = glassy carbon.
Another form of metal-free in-situ bromine generation is the use of HBr as a bromine source. Lei et al. reported an electrochemical oxidative clean halogenation of alkenes using HBr on a carbon anode and platinum cathode, providing 23 examples with up to 89% isolated yields under constant current conditions [40]. The method provides a general halogenation system of HX/MX including the chlorination and bromination of various heteroarenes, arenes, alkenes, aliphatic hydrocarbons and alkynes as well. Under the optimal conditions, the gram-scale synthesis of the electrochemical dibromination of dodecane (22) allowed the formation of 1,2-dibromododecane (23) with a yield of 86% and 50.9 g isolated clean product (Scheme 10).
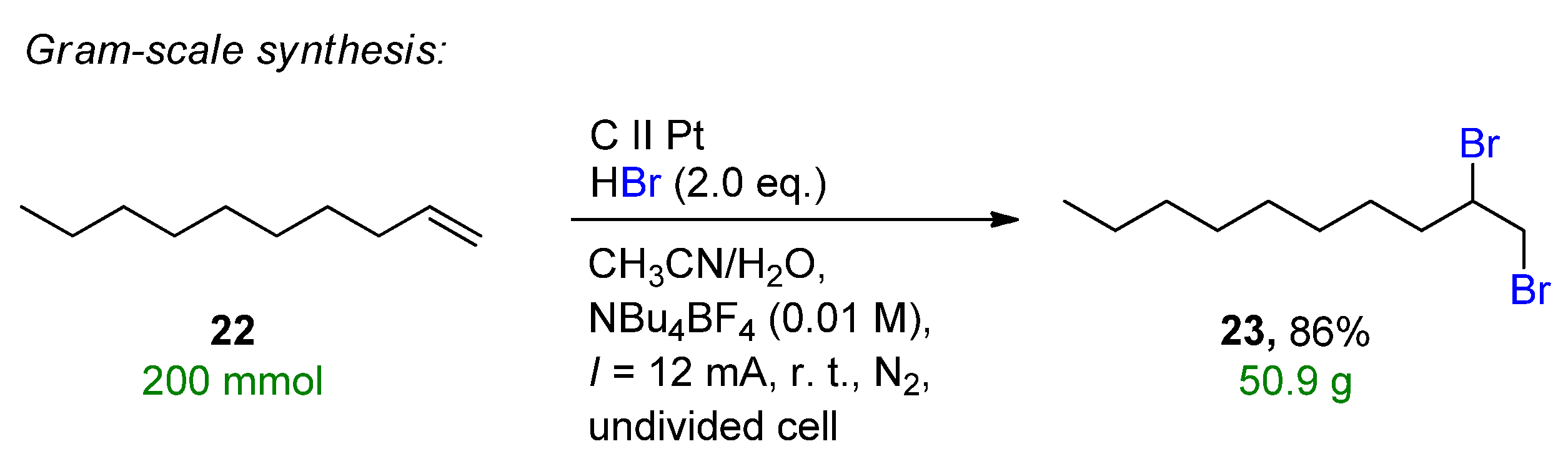
Scheme 10. Electrochemical dibromination of 22 on a gram scale [40]. CCE conditions were described with current (I), as the immersed electrode area was not specified.
Encouraged by these results, the Wirth group described the electrochemical bromination and bromofunctionalization of activated and unactivated alkenes in a flow reactor under single-pass conditions [41]. The 600 µL reactor was equipped with platinum foil electrodes separated by a 500 µM fluorinated ethylene propilene (FEP) spacer. The small interelectrode distance allowed the omission of the addition of water and supporting electrolyte, which was described in previous studies. The optimized conditions for the dibromination of styrene were found in the presence of 6 eq. HBr in pure acetonitrile. The combination of a 0.4 mL/min flow rate, 4 F applied charge and platinum electrodes allowed the formation of 25a in a 79% isolated yield (Scheme 11). When the optimized conditions were applied using a platinum-coated titanium cathode, 25a could be obtained in an excellent yield of 86%. Via switching the co-solvent, the electrochemical bromohydrin formation could be also targeted. The applicability of this method was demonstrated on several aromatic and aliphatic alkenes. In total, 33 examples were provided exhibiting the formation of dibrominated, tetrabrominated, hydrobrominated and alkoxybrominated products with good to excellent yields. The scalability of the flow-procedure was also demonstrated via applying the conditions for 9.5 h providing 25a in an isolated yield of 65% and a productivity of 413 mg/h.
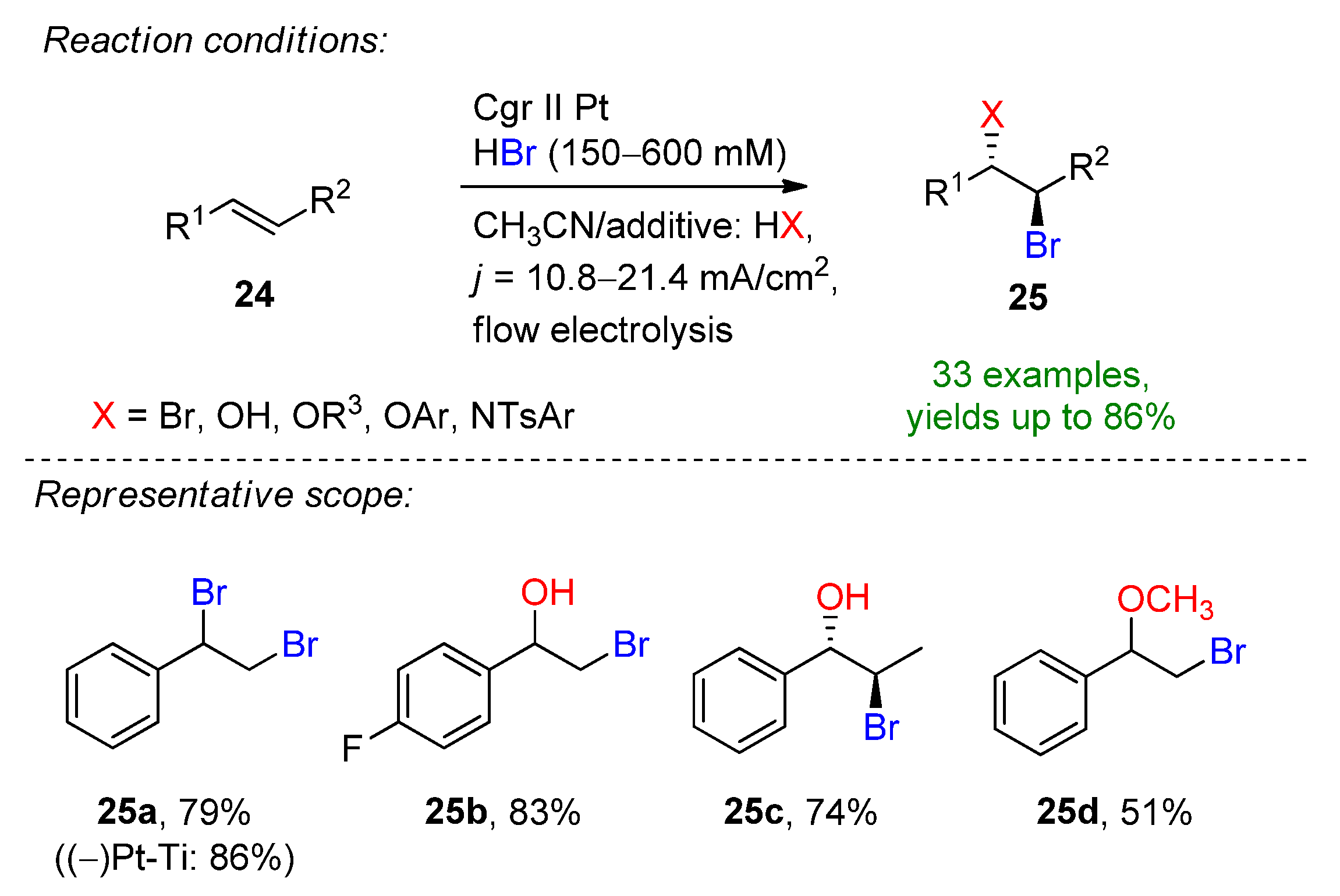
Scheme 11. Electrochemical bromination and bromofunctionalization of alkenes in flow electrolysis [41]. Cgr = graphite.
2.2. Organohalides as Bromine Sources
Li et al. reported a three-component, TEMPO-mediated 1,2-bromoesterification of alkenes with the aid of carboxylic acids and N-bromosuccinimide [42]. The method allows the simultaneous addition of a C–O and C–Br bond to form β-bromoalkyl esters, which are excellent intermediates for natural products and pharmaceutical agents [42][43]. The reaction proceeded well in the presence of 50 mol% TEMPO, 2 eq. of carboxylic acids and 2 eq. NBS to form 27b in an 88% isolated yield (Scheme 12). The optimized conditions were extended to a variety of carboxylic acids and alkenes, and a total of 40 examples were provided with excellent yields. Even challenging substrates such as adamantane-1-carboxylic acid and amino acids were tolerated. The reaction is complemented via H2 evolution at the cathode.
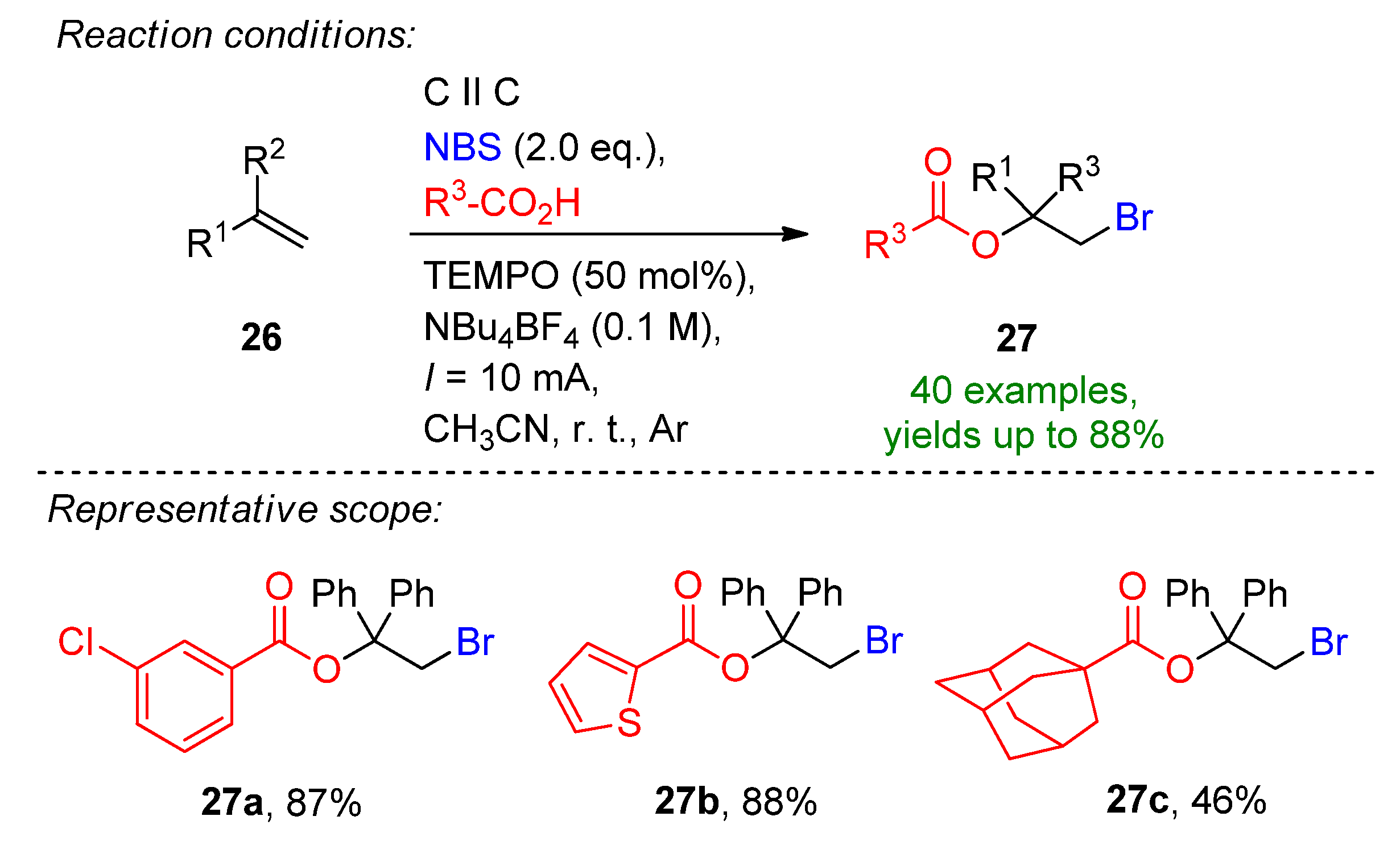
Scheme 12. The three-component, TEMPO-mediated 1,2-bromoesterification of alkenes [42]. CCE conditions were described with current (I), as the immersed electrode area was not specified.
Paired electrolysis methods are generally preferred over a sacrificial approach [44]. Here, the same authors introduced electrochemical alkoxyhalogenation and organohalide dehalogenation within a convergent strategy [45]. Diethyl-2-bromomalonate (29) serves as the bromine source. The key promoters of this method are NBu4OH and Cp2Fe, as omitting one or both decreases the yield. The reaction mechanism was postulated via the mediated oxidation of alkene 28 by Cp2Fe+, which forms the radical cation IX. Intermediate IX is scavenged by the bromine radical or bromine that is either formed via the cathodic reduction of bromomalonate to XIII or via the SN2 reaction induced by the supporting electrolyte (Scheme 13).
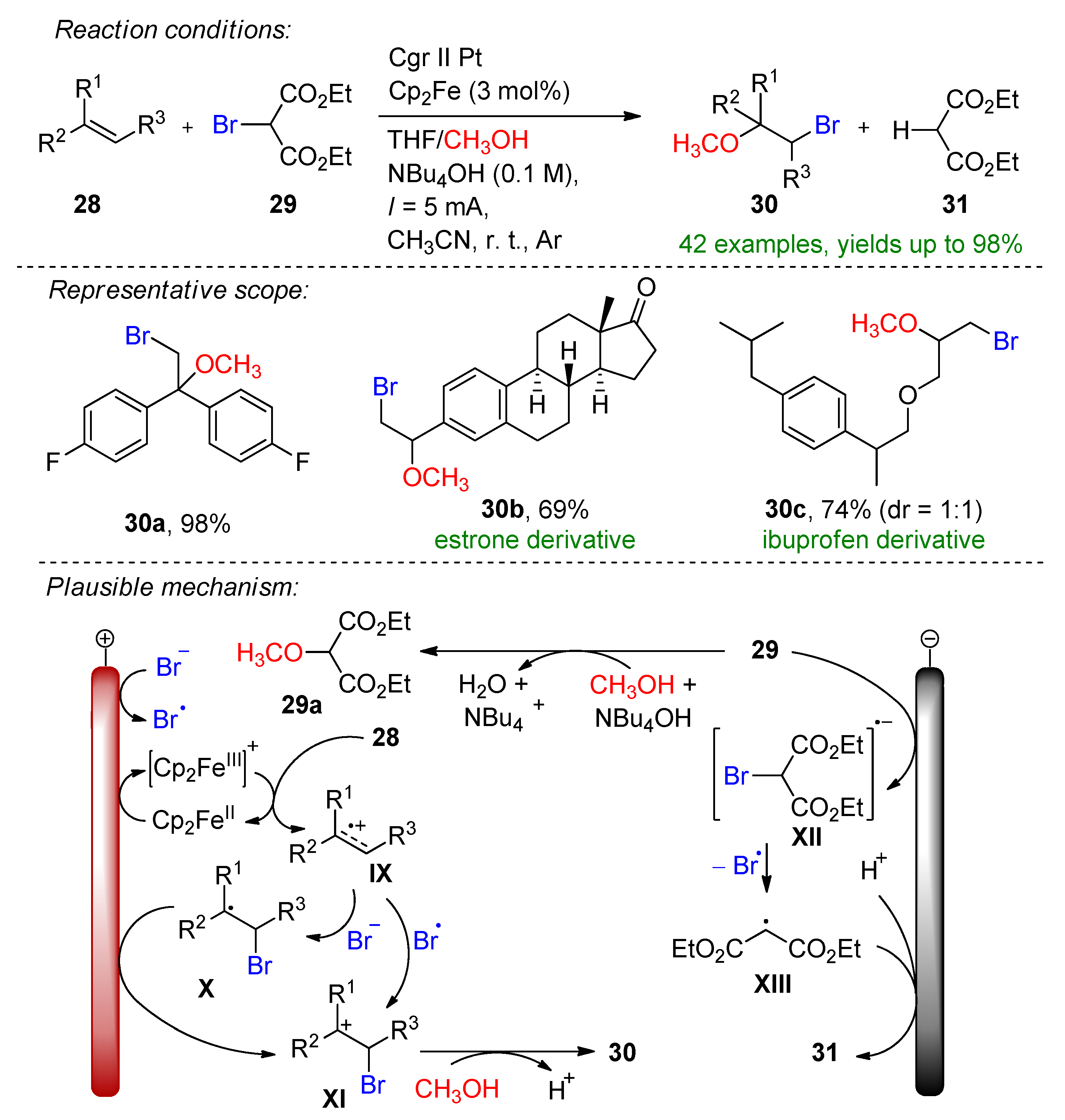
Scheme 13. The electrochemical alkoxyhalogenation of alkenes within a convergent paired electrolysis method [45]. CCE conditions were described with current (I), as the immersed electrode area was not specified. Cgr = graphite.
The bromide could also be oxidized in-situ to form bromine. The formed alkyl cation XI is complemented via methanol to provide the desired product. The formation of malonic ester (31) is depicted via GC and GC-MS. The convergent strategy was demonstrated on various aryl alkenes bearing para-, meta- or both substituents and alkyl halides. The alkoxyhalogenation of bioactive molecules such as adamantene, estrone and ibuprofen derivatives were also demonstrated. In combination, 43 examples were provided with up to a 98% yield.
A merging e-shuttle reaction for the retro-dihalogenation reaction was developed by Waldvogel and Morandi, enhancing the already existing halogenation reactions (Scheme 14) [46].
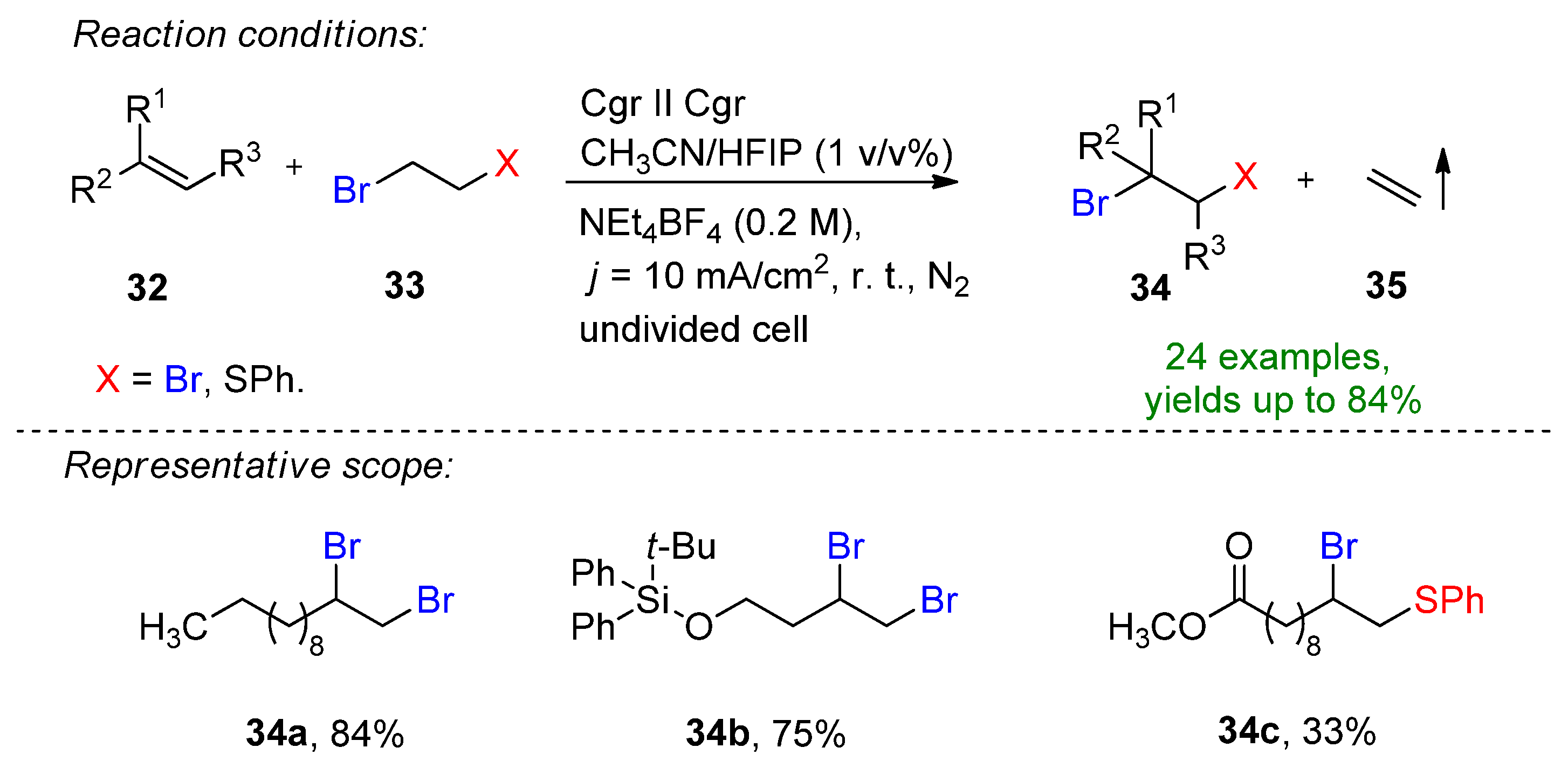
Scheme 14. Merging e-shuttle for the retro-dibromination and bromofunctionalization of alkenes [46]. Cgr = graphite.
References
- Hoegh-Guldberg, O.; Jacob, D.; Taylor, M. Global Warming of 1.5 °C. Available online: https://www.ipcc.ch/sr15/ (accessed on 25 August 2022).
- Yan, M.; Kawamata, Y.; Baran, P.S. Synthetic organic electrochemical methods since 2000: On the verge of a renaissance. Chem. Rev. 2017, 117, 13230–13319.
- Shatskiy, A.; Lundberg, H.; Kärkäs, M.D. Organic electrosynthesis: Applications in complex molecule synthesis. ChemElectroChem 2019, 6, 4067–4092.
- Hilt, G. Basic strategies and types of applications in organic electrochemistry. ChemElectroChem 2020, 7, 395–405.
- Pollok, D.; Waldvogel, S.R. Electro-organic synthesis—A 21st century technique. Chem. Sci. 2020, 11, 12375–12592.
- Novaes, L.F.T.; Liu, J.; Shen, Y.; Lu, L.; Meinhardt, J.M.; Lin, S. Electrocatalysis as an enabling technology for organic synthesis. Chem. Soc. Rev. 2021, 50, 7941–8002.
- Beil, S.B.; Pollok, D.; Waldvogel, S.R. Reproducibility in electroorganic synthesis—Myths and misunderstandings. Angew. Chem. Int. Ed. 2021, 60, 14750–14759.
- Yoshida, J.; Shimizu, A.; Hayashi, R. Electrogenerated cationic reactive intermediates: The pool method and further advances. Chem. Rev. 2017, 118, 4702–4730.
- Torii, S.; Uneyama, K.; Ueda, K. Electrochemical procedure for a practical preparation of piperonal from isosafrole. J. Org. Chem. 1984, 49, 1830–1832.
- Uneyama, K.; Masatsugu, Y.; Torii, S. Electrochemical epoxidation and carbon-carbon bond cleavage for the preparation of 3-methyl-4-oxo-2-phenyl-4H-1-benzopyran-8-carboxylic acid from 3-methyl-2-phenyl-8-(1-prophenyl)-4H-1-benzopyran-4-one. Bull. Chem. Soc. Jpn. 1985, 58, 2361–2365.
- Inokuchi, T.; Matsumoto, S.; Tsuji, M.; Torii, S. Electrohalogenation of propargyl acetates and amides to form the 1,1-dibromo-2-oxo functionality and a facile synthesis of furaneol. J. Org. Chem. 1992, 57, 5023–5027.
- Inês, M.; Mendonça, A.J.; Esteves, A.P.; Mendonça, D.I.; Medeiros, M.J. Electroepoxidation of natural and synthetic alkenes mediated by sodium bromide. Comptes Rendus Chim. 2009, 12, 841–849.
- Kulangiappar, K.; Ramaprakash, M.; Vasudevan, D.; Raju, T. Electrochemical bromination of cyclic and acyclic enes using biphasic electrolysis. Synth. Commun. 2016, 46, 145–153.
- Yang, C. An impending platinum crisis and its implications for the future of the automobile. Energy Policy 2009, 37, 1805–1808.
- Das, K.K.; Reddy, R.C.; Bagoji, I.B.; Das, S.; Bagali, S.; Mullur, L.; Khodnapur, J.P.; Biradar, M.S. Primary concept of nickel toxicity—An overview. J. Basic Clin. Physiol. Pharmacol. 2019, 30, 141–152.
- Savignan, L.; Faucher, S.; Chéry, P.; Lespes, G. Platinum group elements contamination in soils: Review of the current state. Chemosphere 2021, 271, 129517.
- Sun, X.; Ma, H.; Mei, T.; Fang, P.; Hu, Y. Electrochemical radical formyloxylation-bromination, -chlorination, and -trifluoromethylation of alkenes. Org. Lett. 2019, 21, 3167–3171.
- Bormann, S.; Van Schie, M.M.C.H.; De Almeida, T.P.; Zhang, W.; Stöckl, M.; Ulber, R.; Hollman, F.; Holtmann, D. H2O2 production at low overpotential for electroenzymatic halogenation reactions. ChemSusChem 2019, 12, 4759–4763.
- Wever, R.; Renirie, R.; Hollmann, F. Vanadium chloroperoxidases as versatile biocatalysts. In Vanadium Catalysis; Sudradhar, M., Pombeiro, A.J.L., da Silva, J.A.L., Eds.; Royal Society of Chemistry: London, UK, 2021; Chapter 24; ISBN 978-1-78801-857-9.
- Wu, J.; Abou-Hamdan, H.; Guillot, R.; Kouklovsky, C.; Vincent, G. Electrochemical synthesis of 3a-bromofuranoindolines and 3a-bromopyrroloindolines mediated by MgBr2. Chem. Commun. 2020, 56, 1713–1716.
- Hakamata, H.; Sato, S.; Ueda, H.; Tokuyama, H. AgNTf2-Mediated Allylation with Allylsilanes at C3a-Position of Hexahydropyrroloindoles: Application to Total Syntheses of Amauromine Alkaloids. Org. Lett. 2017, 19, 5308–5311.
- Ramos-Villaseñor, J.M.; Rodríguez-Cárdenas, E.; Díaz, C.E.B.; Frontana-Uribe, B.A. Review—Use of 1,1,1,3,3,3–hexafluoro–2–propanol (HFIP) Co-Solvent Mixtures in Organic Electrosynthesis. J. Electrochem. Soc. 2020, 167, 155509.
- Eberson, L.; Hartshorn, M.P.; Persson, O. 1,1,1,3,3,3-Hexafluoropropan-2-ol as a solvent for the generation of highly persistent radical cations. J. Chem. Soc. Perkin Trans. 2 1995, 1735–1744.
- Eberson, L.; Hartshorn, M.P.; Persson, O. Generation of solutions of highly persistent radical cations by 4-tolylthallium(III) bis(trifluoroacetate) in 1,1,1,3,3,3-hexafluoropropan-2-ol. J. Chem. Soc. Chem. Commun. 1995, 1131–1132.
- Eberson, L.; Persson, O.; Hartshorn, M.P. Detection and Reactions of Radical Cations Generated by Photolysis of Aromatic Compounds with Tetranitromethane in 1,1,1,3,3,3-Hexafluoro-2-propanol at Room Temperature. Angew. Chem. Int. Ed. 1995, 34, 2268–2269.
- Schulz, L.; Waldvogel, S.R. Solvent control in electro-organic synthesis. Synlett 2018, 30, 275–286.
- Röckl, J.L.; Dörr, M.; Waldvogel, S.R. Electrosynthesis 2.0 in 1,1,1,3,3,3-Hexafluoroisopropanol/Amine Mixtures. ChemElectroChem 2020, 7, 3686–3694.
- Ashikari, Y.; Shimizu, A.; Nokami, T.; Yoshida, J. Halogen and Chalcogen Cation Pools Stabilized by DMSO. Versatile Reagents for Alkene Difunctionalization. J. Am. Chem. Soc. 2013, 135, 16070–16073.
- Shimizu, A.; Hayashi, R.; Ashikari, Y.; Nokami, T.; Yoshida, J. Switching the reaction pathways of electrochemically generated β-haloalkoxysulfonium ions—Synthesis of halohydrins and epoxides. Beilstein J. Org. Chem. 2015, 11, 242–248.
- Bityukov, O.V.; Vil’, V.A.; Nikishin, G.I.; Terent’ev, A.O. Alkene, Bromide, and ROH—How To Achieve Selectivity? Electrochemical Synthesis of Bromohydrins and Their Ethers. Adv. Synth. Catal. 2021, 363, 3070–3078.
- Shang, X.; Liu, X.; Sun, Y. Flexible on-site halogenation paired with hydrogenation using halide electrolysis. Green Chem. 2021, 23, 2037–2043.
- Strehl, J.; Abraham, M.L.; Hilt, G. Linear Paired Electrolysis—Realising 200 % Current Efficiency for Stoichiometric Transformations—The Electrochemical Bromination of Alkenes. Angew. Chem. Int. Ed. 2021, 60, 9996–10000.
- Pitzer, L.; Schäfers, F.; Glorius, F. Rapid Assessment of the Reaction-Condition-Based Sensitivity of Chemical Transformations. Angew. Chem. Int. Ed. 2019, 58, 8572–8576.
- Ye, J.H.; Quach, L.; Paulisch, T.; Glorius, F. Visible-light-induced, metal-free carbene insertion into B-H bonds between acylsilanes and pinacolborane. J. Am. Chem. Soc. 2019, 141, 16227–16231.
- Kale, A.P.; Nikolaienko, P.; Smirnova, K.; Rueping, M. Intramolecular Electrochemical Oxybromination of Olefins for the Synthesis of Isoxazolines in Batch and Continuous Flow. Eur. J. Org. Chem. 2021, 2021, 3496–3500.
- He, Y.; Qin, X.; He, X.; Wu, X.; Yin, Z. Practical Synthesis of Halogenated N-Heterocycles via Electrochemical Anodic Oxidation of Unactivated Alkenes. Eur. J. Org. Chem. 2021, 2021, 5831–5834.
- Kim, R.; Ha, J.; Woo, J.; Kim, D.Y. Electrochemical oxidative bromolactonization of unsaturated carboxylic acids with sodium bromide: Synthesis of bromomethylated γ-lactones. Tetrahedron Lett. 2022, 88, 153567.
- Yu, D.; Ji, R.; Sun, Z.; Li, W.; Liu, Z. Electrochemical chlorination and bromination of electron-deficient C-H bonds in quinones, coumarins, quinoxalines and 1,3-diketones. Tetrahedron Lett. 2021, 86, 153514.
- Gombos, L.G.; Werner, L.; Schollmeyer, D.; Martínez-Huitle, C.A.; Waldvogel, S.R. Selective Electrochemical Dibromination of Terpenes and Naturally Derived Olefins. Eur. J. Org. Chem. 2022. accepted.
- Yuan, Y.; Yao, A.; Zheng, Y.; Gao, M.; Zhou, Z.; Qiao, J.; Hu, J.; Ye, B.; Zhao, J.; Wen, H.; et al. Electrochemical Oxidative Clean Halogenation Using HX/NaX with Hydrogen Evolution. iScience 2019, 12, 293–303.
- Seitz, J.; Wirth, T. Electrochemical bromofunctionalization of alkenes in a flow reactor. Org. Biomol. Chem. 2021, 19, 6892–6896.
- Wan, C.; Song, R.; Li, J.H. Electrooxidative 1,2-Bromoesterification of Alkenes with Acids and N-Bromosuccinimide. J. Org. Lett. 2019, 21, 2800–2803.
- Pan, G.; Ouyang, X.; Hu, M.; Xie, Y.; Li, J. Copper-Catalyzed Intermolecular Aminoalkylation of Alkenes with α-Bromoalkyl Esters and Amines toward Pyrrolidin-2-ones. Adv. Synth. Catal. 2017, 359, 2564–2570.
- Klein, M.; Waldvogel, S.R. Counter electrode reactions—Important stumbling blocks on the way to a working electro-organic synthesis. Angew. Chem. Int. Ed. 2022. accepted.
- Zhang, T.; Luo, M.; Li, Y.; Song, R.; Li, J. Electrochemical Alkoxyhalogenation of Alkenes with Organohalides as the Halide Sources via Dehalogenation. Org. Lett. 2020, 22, 7250–7254.
- Dong, X.; Roeckl, J.L.; Waldvogel, S.R.; Morandi, B. Merging shuttle reactions and paired electrolysis for reversible vicinal dihalogenations. Science 2021, 371, 507–514.
More
Information
Subjects:
Electrochemistry
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.5K
Revisions:
2 times
(View History)
Update Date:
17 Nov 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No