Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Rita Xu and Version 1 by Lilla Gombos.
The electrochemical generation of highly reactive and hazardous bromine under controlled conditions as well as the reduction of surplus oxidizers and reagent waste has placed electrochemical synthesis in a highlighted position. In particular, the electrochemical dibromination and bromofunctionalization of alkenes and alkynes have received significant attention, as the forming of synthetically important derivatives can be generated from bench-stable and safe bromide sources under “green” conditions.
- anode
- alkenes
- bromine
- electrochemistry
1. Introduction
With the increasing amount of economic, environmental and political pressure, the chemical industry faces the challenge of providing the public and the scientific community a sustainable and greener alternative to conventional chemical methods [1]. Electro-organic synthesis has become one of the most attractive research topics in recent years, providing highly versatile synthetic methodologies [2,3,4,5,6][2][3][4][5][6]. Using electrons as “traceless” redox reagents allows the elimination of the excess use of harmful redox chemicals with a high atom efficiency [7]. The electro-generation of highly reactive species in-situ allows the utilization of inherently safe surrogates and provides a simplified approach [8]. These advantages, in combination with the valorization of renewable feedstock and “green electricity”, makes electrochemical functionalization particularly attractive.
2. Electrochemical Bromofunctionalization of Alkenes
2.1. HBr, MBr and Alkylammonium Bromides as Bromine Sources
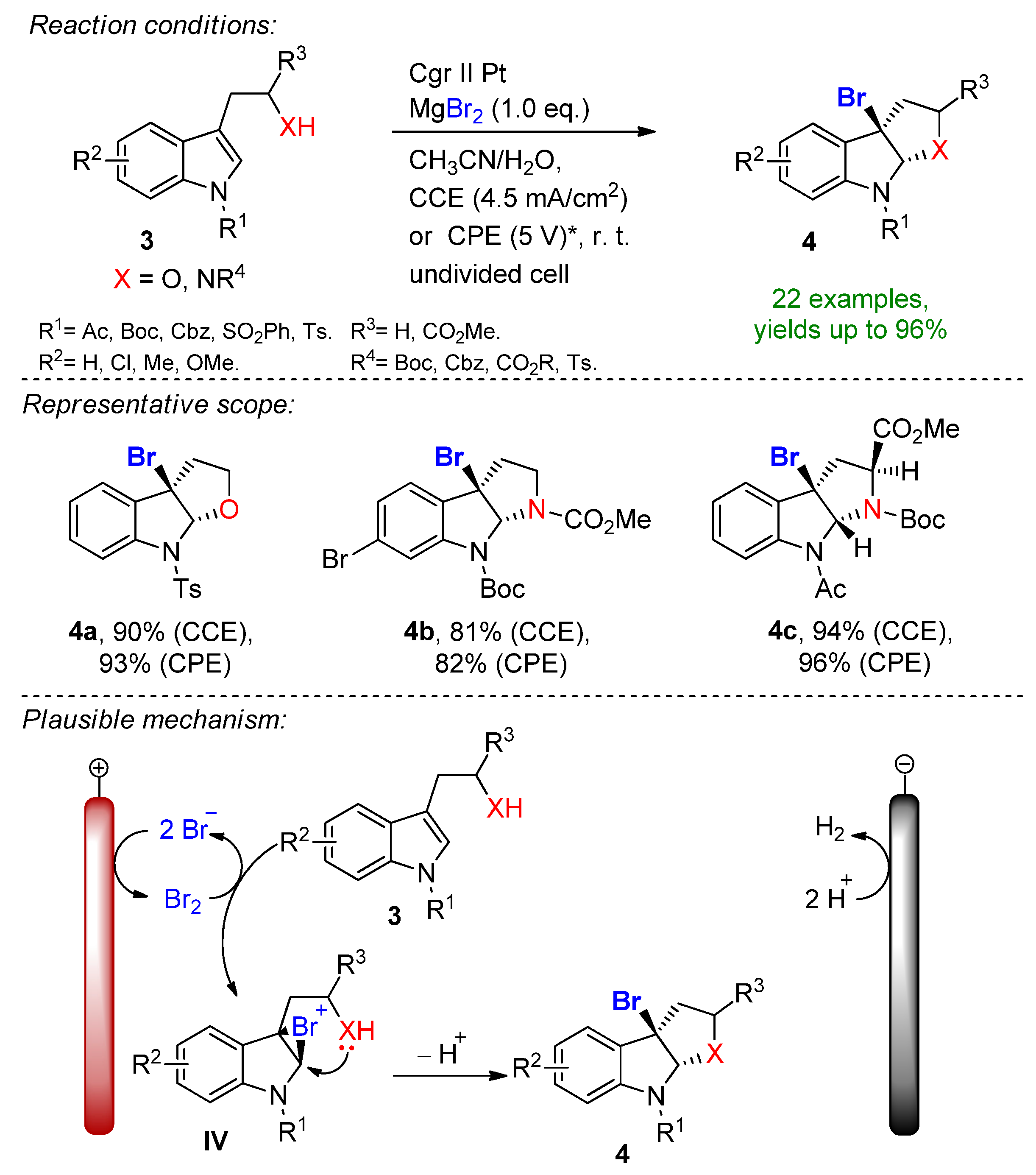
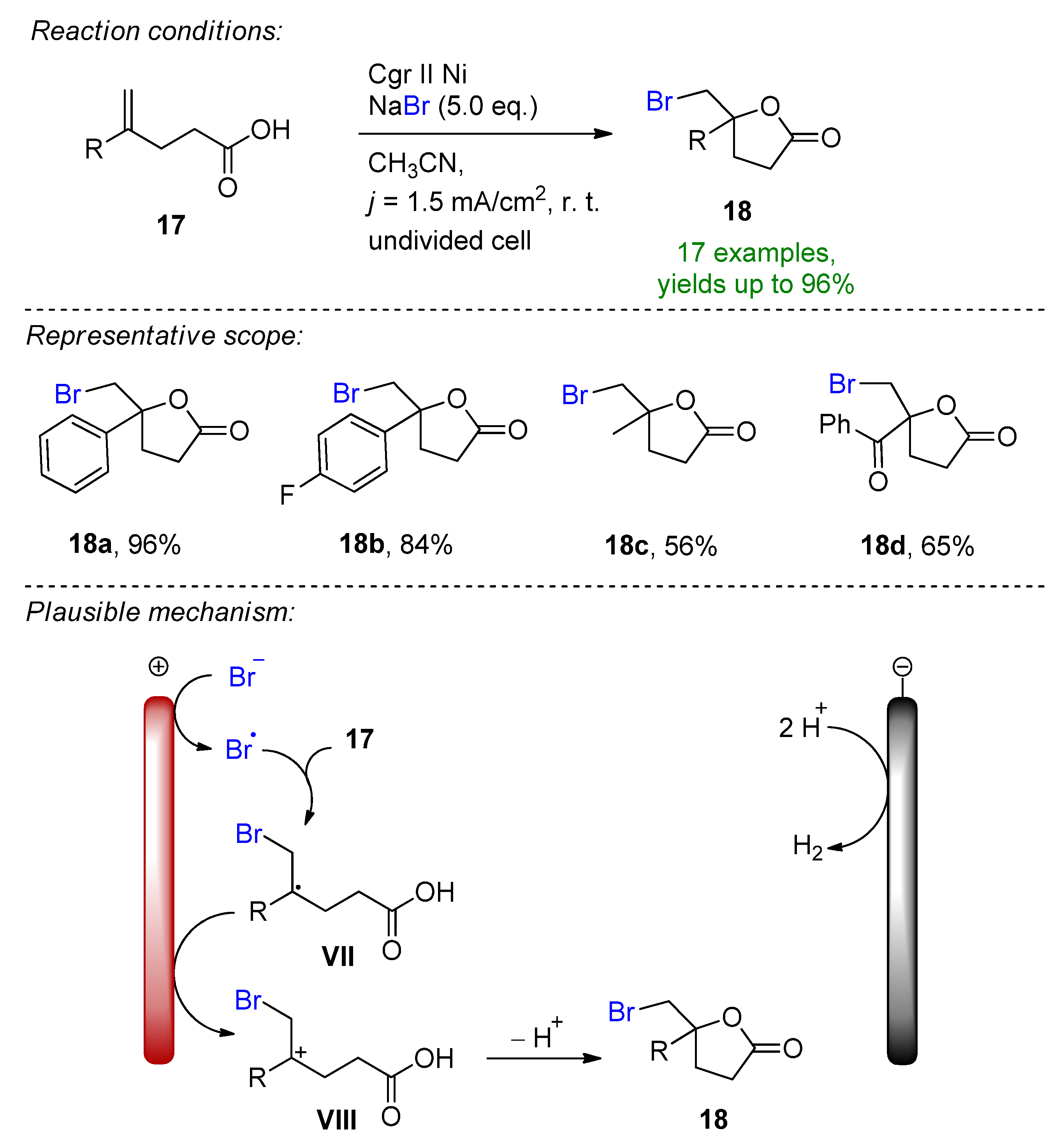
Pioneering work in the field was demonstrated by Torii in the early 80s, reporting the bromofunctionalization of alkenes in the presence of aq. NaBr, which popularized the indirect electrochemical oxidation of olefins using stable and inexpensive metal halide salts [64,65,66,67,68][9][10][11][12][13]. This form of a bromine source has received particular attention due to its ability to tame hazardous and toxic molecular bromine in-situ under controlled conditions. It is worth mentioning that the complementary half-reaction´s electrode choice is usually a low-hydrogen-overpotential material such as platinum or nickel, which supports cathodic hydrogen evolution. Nevertheless, platinum group metals are depleting, costly and highly contaminating [69,70,71][14][15][16]. In 2019 Hu, Fang and Mei reported the electrochemical radical formyloxylation–bromination of various alkenes in a regio- and chemoselective fashion [72][17]. The implementation of stable NaBr as a radical bromine source makes this approach sustainable. DMF serves a dual role as both the solvent and the formyloxylation reagent, while acetic acid supports the cathodic reaction. Para-substituted electron-rich styrene derivatives afforded excellent yields, while electron-deficient and strongly electron-withdrawing groups prolonged the reaction time. The naturally derived safrole-type and estrone-type derivates also readily underwent electrochemical formyloxylation, proving the possibility for the late-stage functionalization of pharmaceutical scaffolds. In total, 30 examples were reported with excellent yields up to 97%. The scalability was proved via a gram-scale experiment (6.0 mmol) resulting in an isolated yield of 90%. The method was extended to the formyloxylation–chlorination and formyloxylation–trifluoromethylation of alkenes as well. The authors suggest that bromide is oxidized at the anode to bromo-radicals, which then rapidly combine with arylalkene 1 to form the benzyl radical I. Via a second oxidation, the forming benzylic carbocation II is subjected to nucleophilic attack by DMF to iminium intermediate III, which is subsequently hydrolyzed resulting in the formyloxylated brominated product 2 (Scheme 1). The electrobiocatalytic bromolactonization was reported by Bormann and Holtmann on carbon-nanotube-modified gas-diffusion electrodes [73][18]. The paper reports the cathodic reduction of ambient oxygen to H2O2 at a reduced overpotential in a divided cell separated via a proton exchange membrane. Curvularia inaequalis (CiVCPO), a vanadium-dependent chloroperoxidase enzyme converts the forming peroxide in the presence of potassium bromide into hypobromite, which leads to the bromolactonization of 5-pentenoic acid. The reaction is complimented via the oxidation of water at a platinum anode. The superiority of the reaction lies in the oxygen-enriched, modified-carbon-nanotube diffusion electrode that significantly lowers the overpotential for peroxide formation, which is a sensible factor for the enzymatic catalytic reactivity [74][19]. The electrochemical bromocyclization of tryptophol, tryptamine and tryptophan derivatives was recently reported by Wu and Vincent [75][20]. This efficient protocol features MgBr2 with a dual role as a halogen source and supporting electrolyte. The reaction proceeds in an undivided cell under both constant current (CCE) and constant potential (CPE) conditions in the presence of 1 eq. MgBr2. Magnesium bromide is oxidized at the carbon anode into bromine in an acetonitrile/water solution under ambient conditions. It is worth mentioning that under CPE, the constant potential window is maintained via measuring the potential against a reference electrode. Here, the constant terminal voltage of the cell was determined via measuring the constant terminal voltage between the electrodes, which is not an adequate parameter for CPE conditions. The dearomative reaction proceeds via the formation of bromonium intermediate IV, followed by intramolecular cyclization to form the corresponding brominated derivatives with excellent yields (Scheme 2). This simple and environmentally friendly set up featured 22 examples with up to 96% isolated yields and a broad scope of functional group tolerance.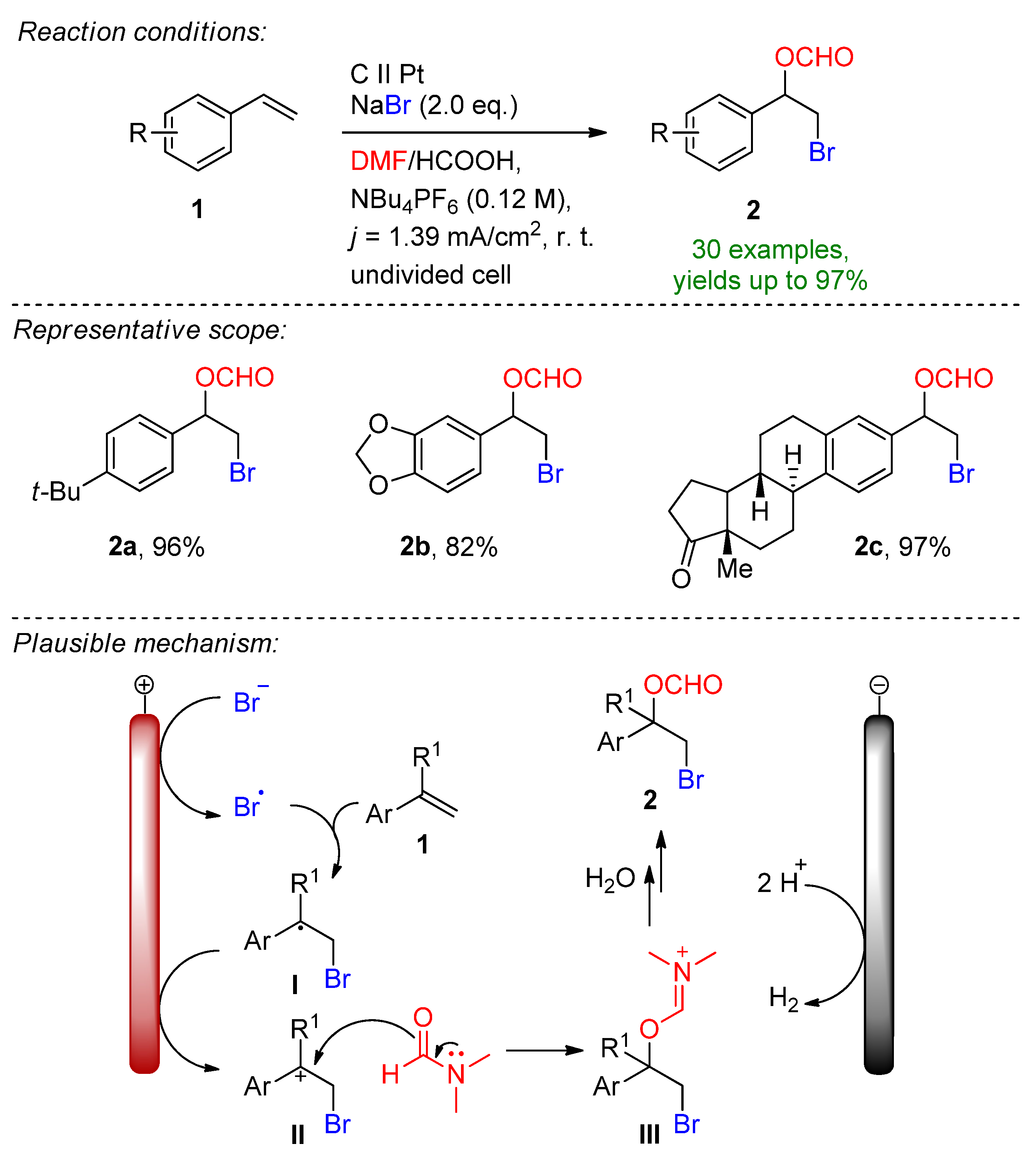
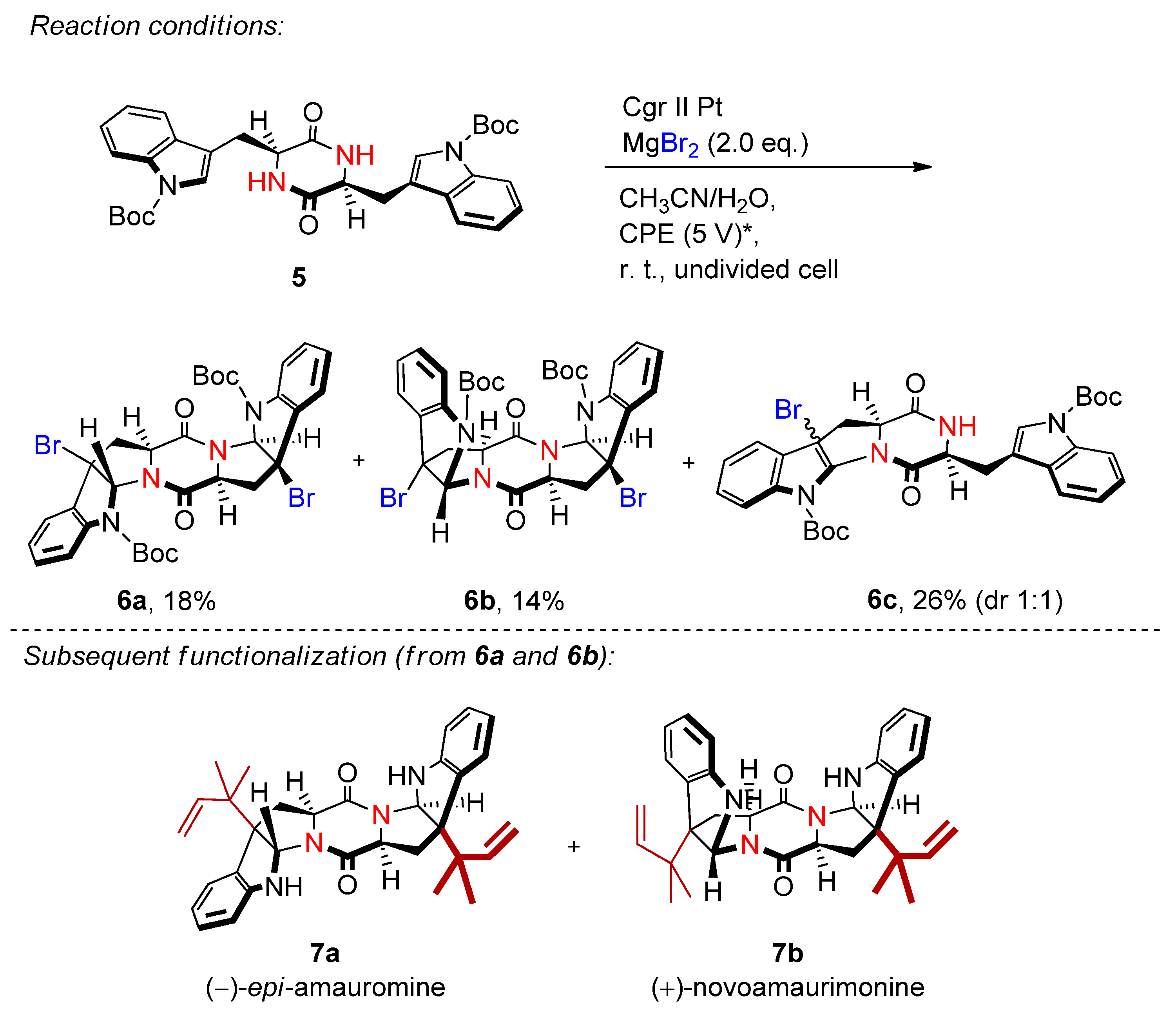
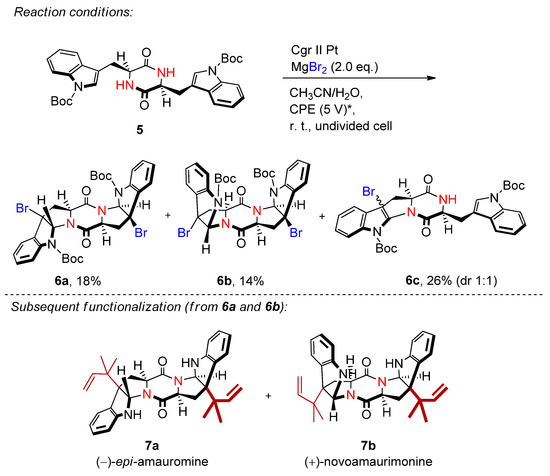
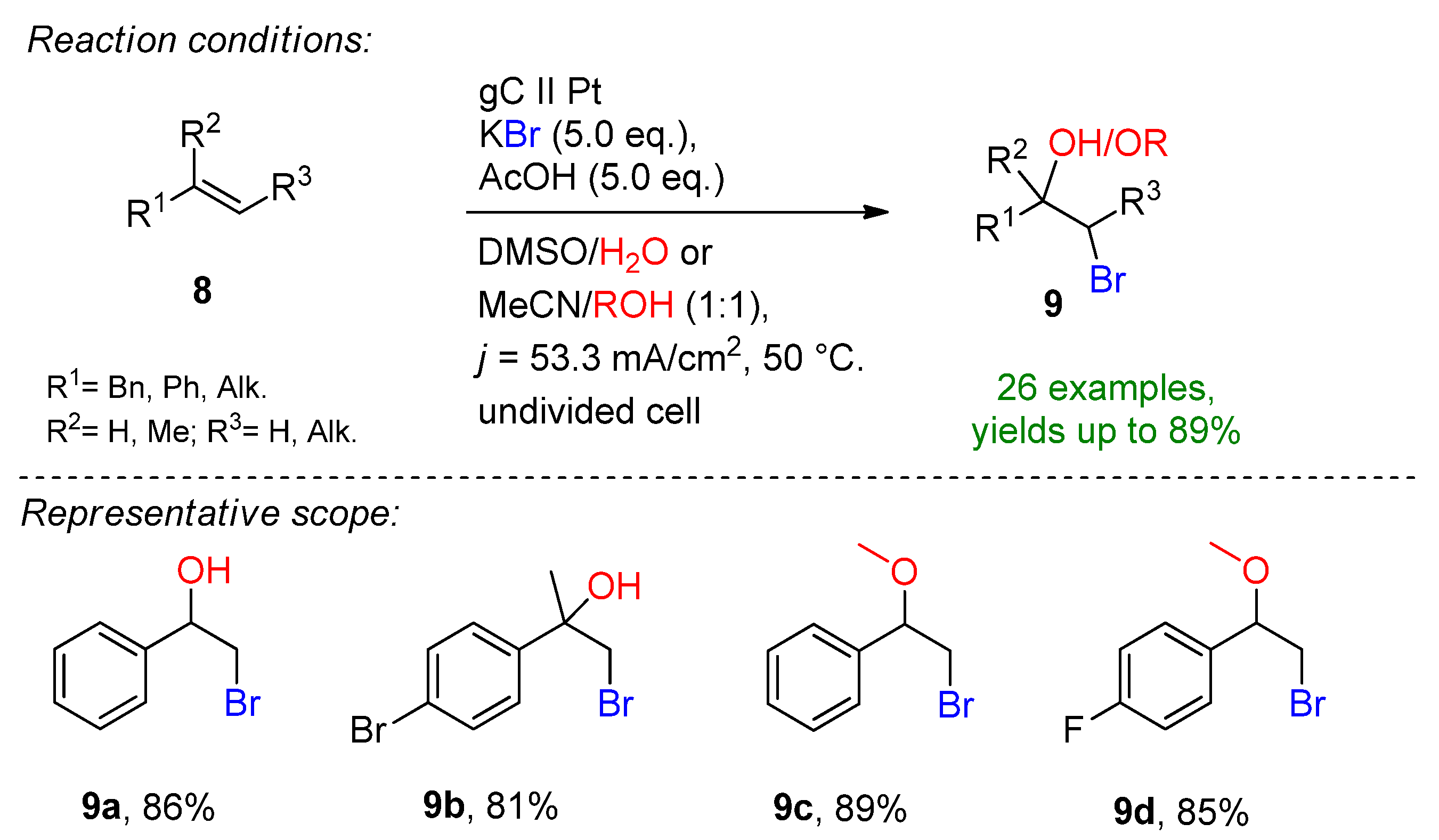
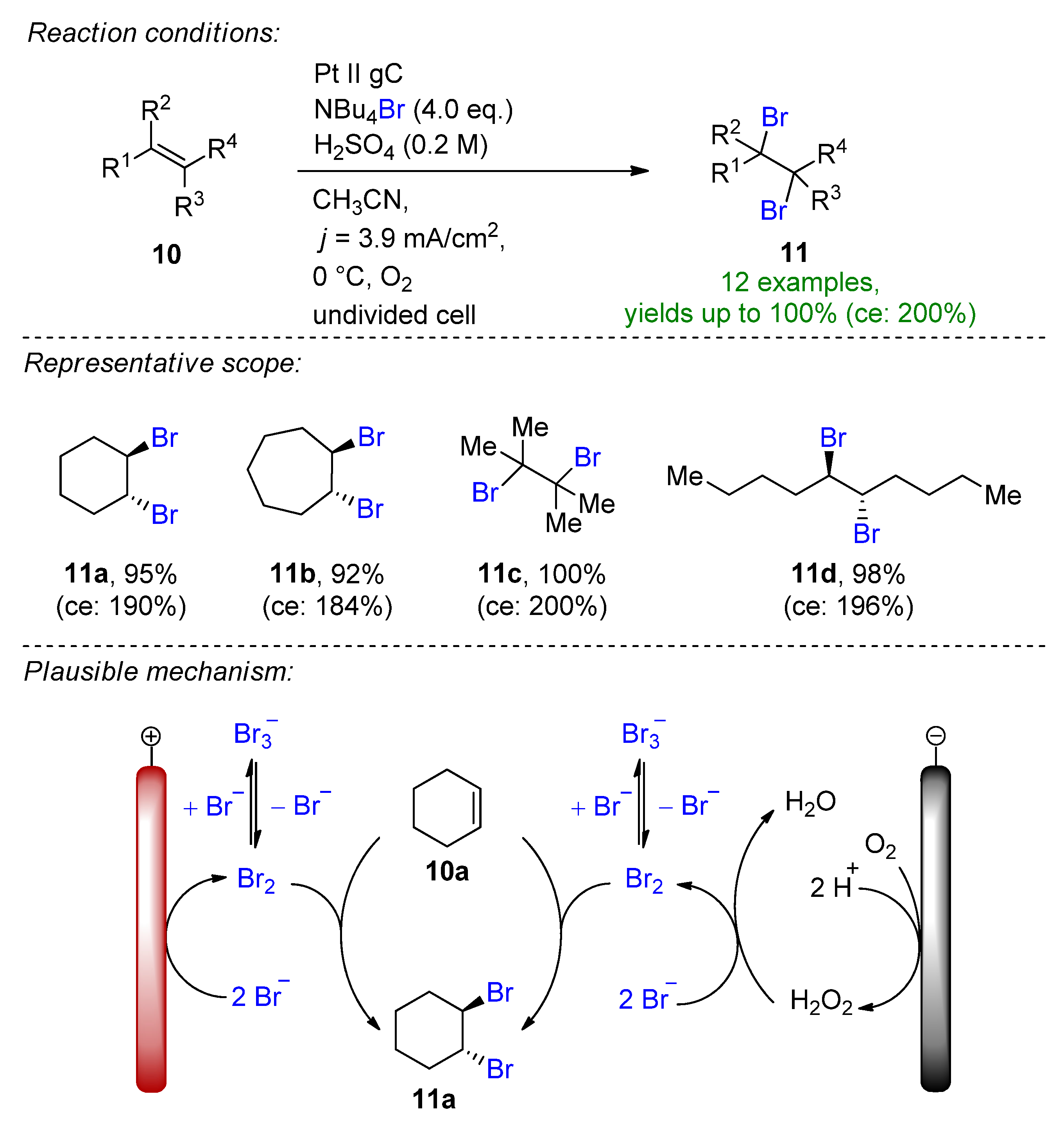
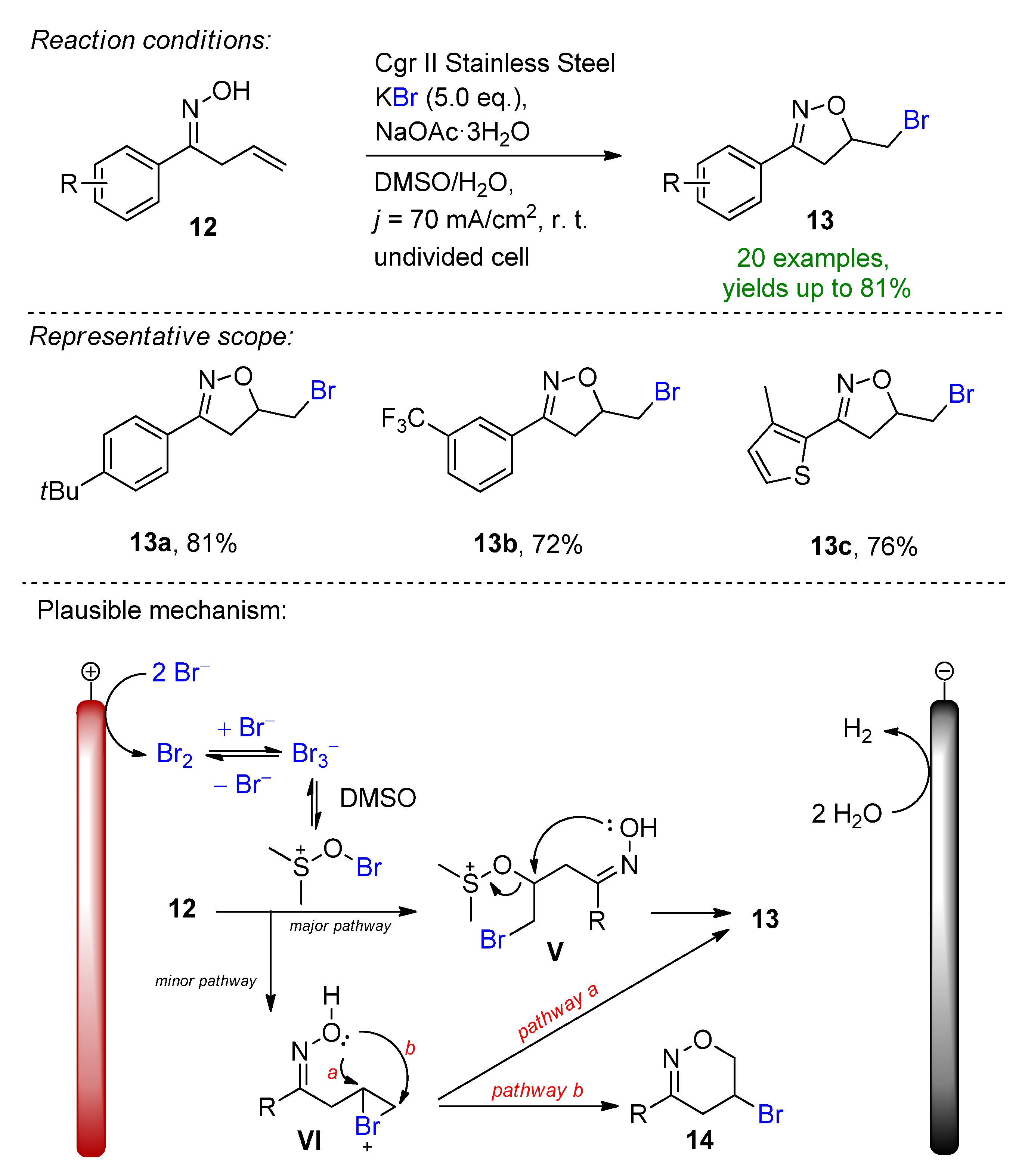
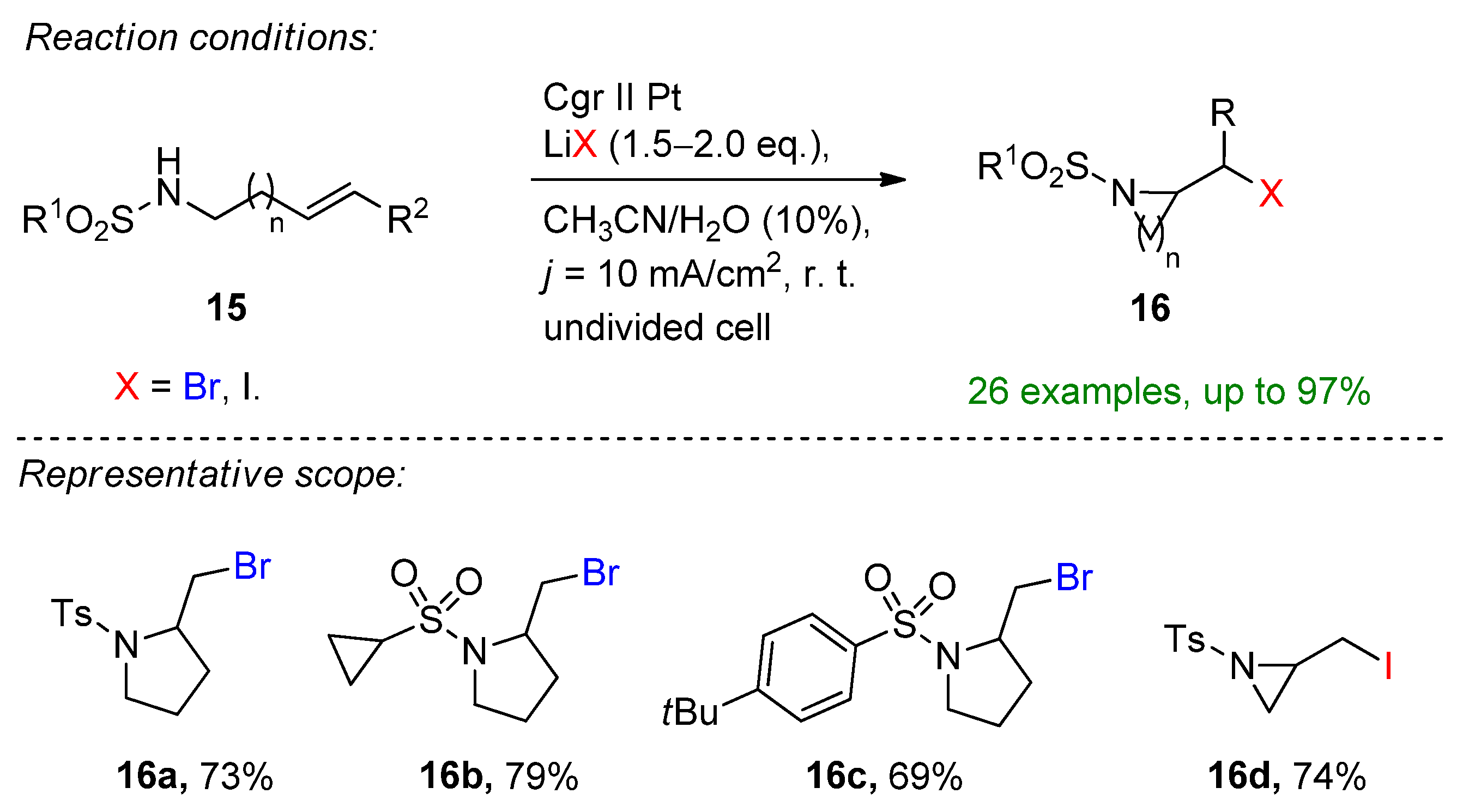
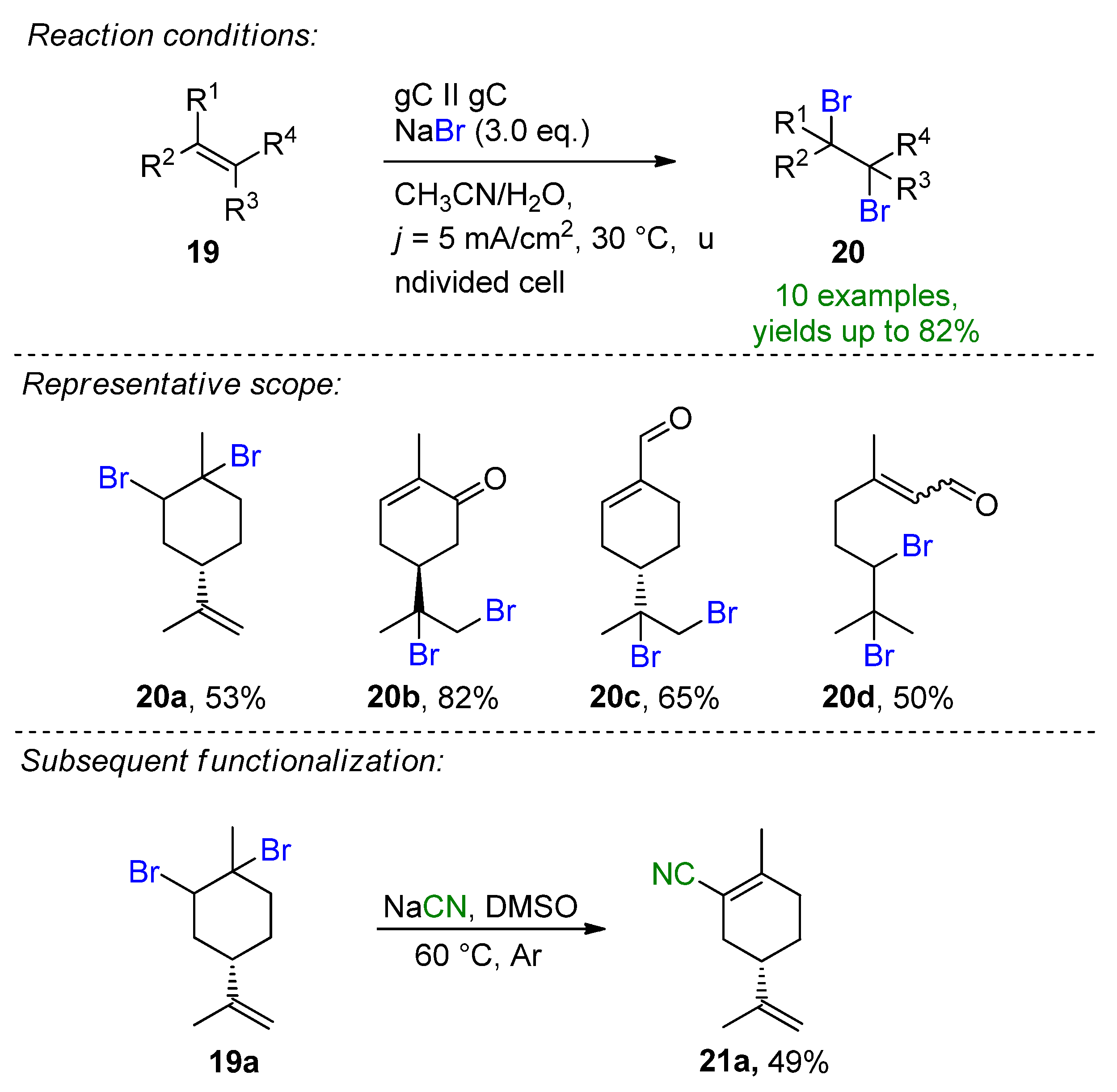
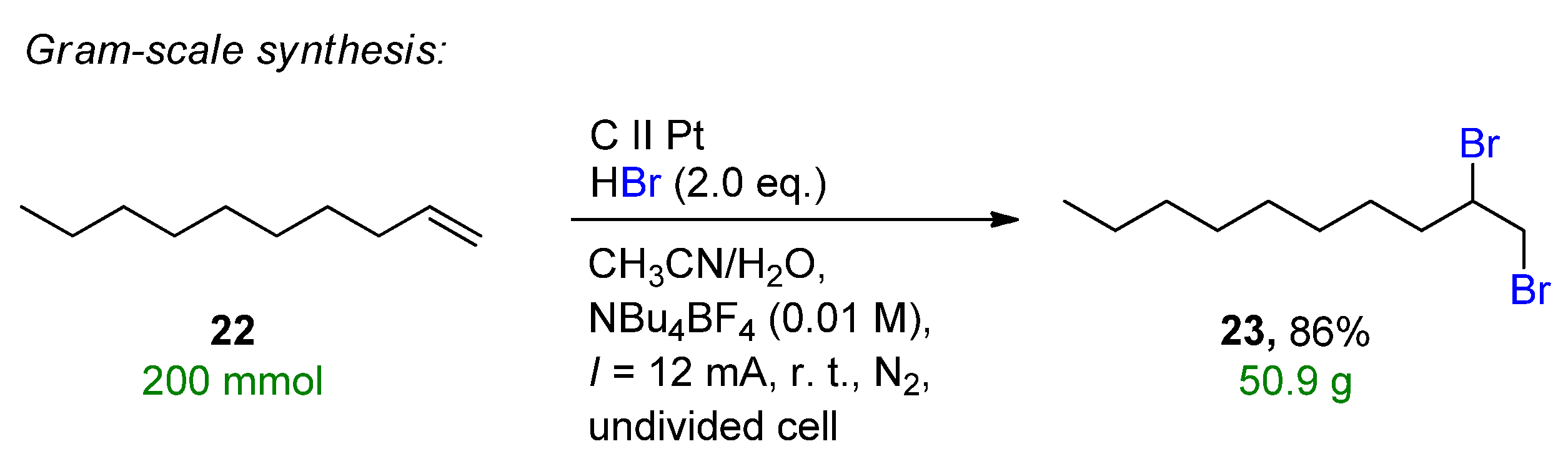
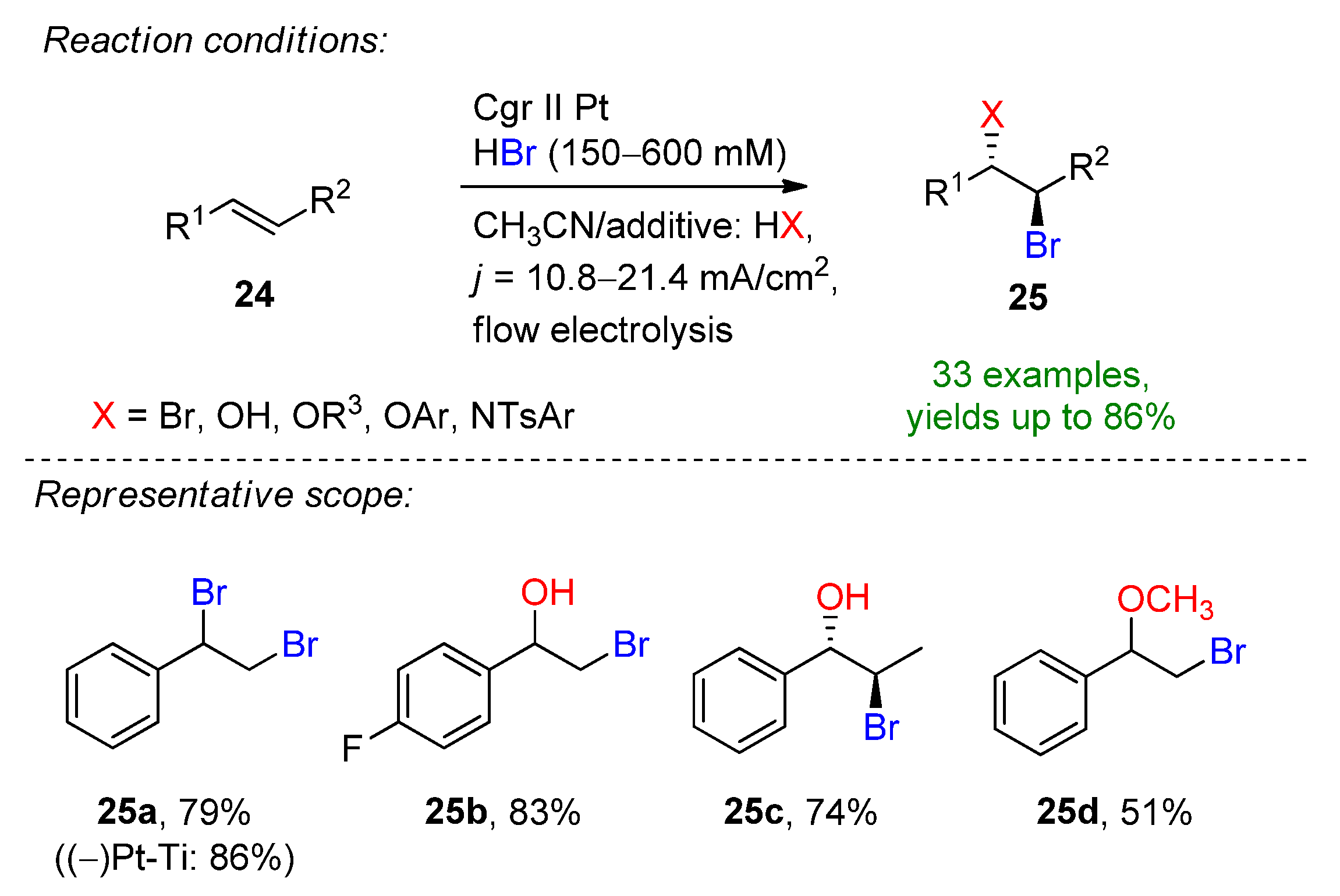
2.2. Organohalides as Bromine Sources
Li et al. reported a three-component, TEMPO-mediated 1,2-bromoesterification of alkenes with the aid of carboxylic acids and N-bromosuccinimide [97][42]. The method allows the simultaneous addition of a C–O and C–Br bond to form β-bromoalkyl esters, which are excellent intermediates for natural products and pharmaceutical agents [97,98][42][43]. The reaction proceeded well in the presence of 50 mol% TEMPO, 2 eq. of carboxylic acids and 2 eq. NBS to form 27b in an 88% isolated yield (Scheme 12). The optimized conditions were extended to a variety of carboxylic acids and alkenes, and a total of 40 examples were provided with excellent yields. Even challenging substrates such as adamantane-1-carboxylic acid and amino acids were tolerated. The reaction is complemented via H2 evolution at the cathode.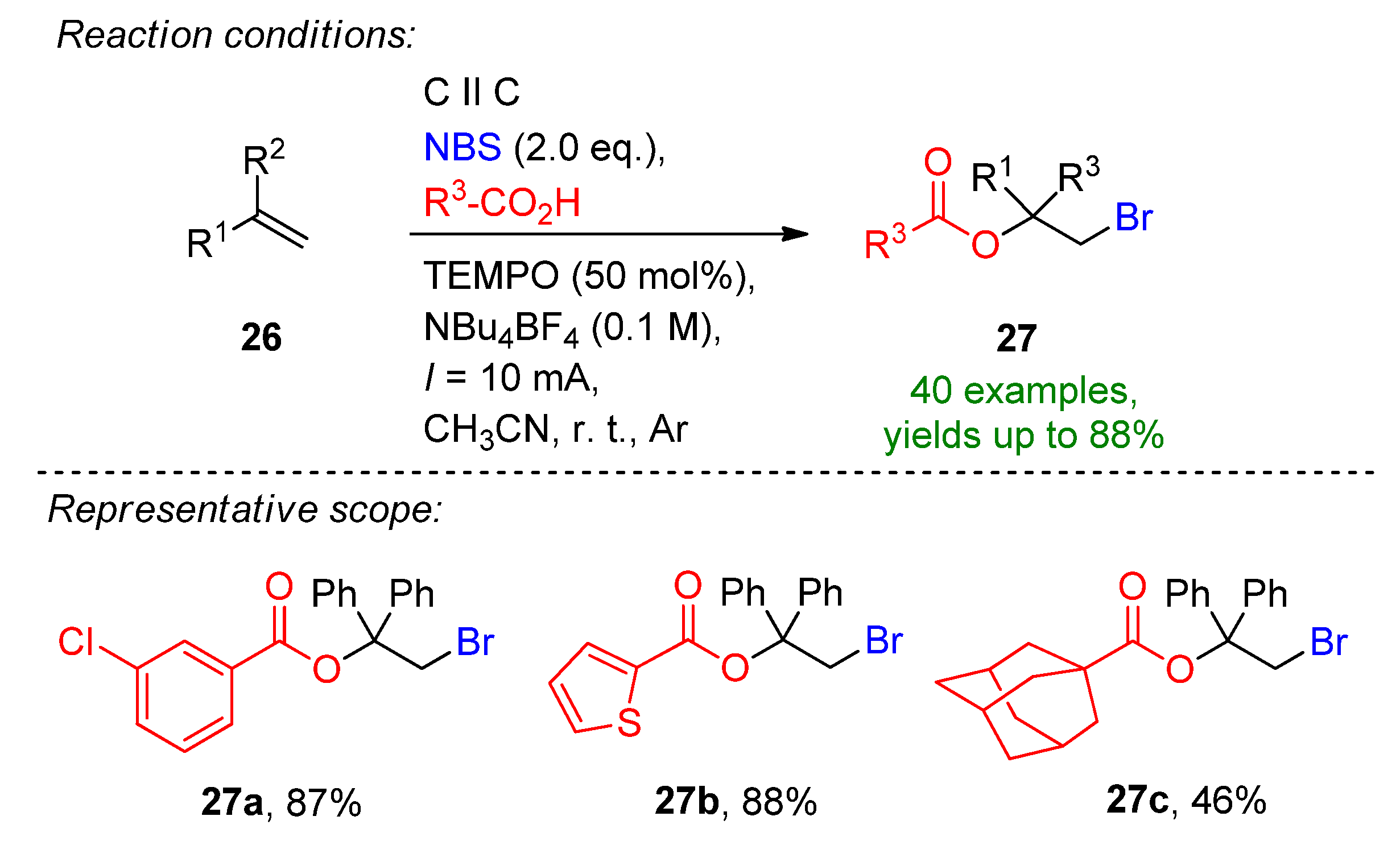
References
- Hoegh-Guldberg, O.; Jacob, D.; Taylor, M. Global Warming of 1.5 °C. Available online: https://www.ipcc.ch/sr15/ (accessed on 25 August 2022).
- Yan, M.; Kawamata, Y.; Baran, P.S. Synthetic organic electrochemical methods since 2000: On the verge of a renaissance. Chem. Rev. 2017, 117, 13230–13319.
- Shatskiy, A.; Lundberg, H.; Kärkäs, M.D. Organic electrosynthesis: Applications in complex molecule synthesis. ChemElectroChem 2019, 6, 4067–4092.
- Hilt, G. Basic strategies and types of applications in organic electrochemistry. ChemElectroChem 2020, 7, 395–405.
- Pollok, D.; Waldvogel, S.R. Electro-organic synthesis—A 21st century technique. Chem. Sci. 2020, 11, 12375–12592.
- Novaes, L.F.T.; Liu, J.; Shen, Y.; Lu, L.; Meinhardt, J.M.; Lin, S. Electrocatalysis as an enabling technology for organic synthesis. Chem. Soc. Rev. 2021, 50, 7941–8002.
- Beil, S.B.; Pollok, D.; Waldvogel, S.R. Reproducibility in electroorganic synthesis—Myths and misunderstandings. Angew. Chem. Int. Ed. 2021, 60, 14750–14759.
- Yoshida, J.; Shimizu, A.; Hayashi, R. Electrogenerated cationic reactive intermediates: The pool method and further advances. Chem. Rev. 2017, 118, 4702–4730.
- Torii, S.; Uneyama, K.; Ueda, K. Electrochemical procedure for a practical preparation of piperonal from isosafrole. J. Org. Chem. 1984, 49, 1830–1832.
- Uneyama, K.; Masatsugu, Y.; Torii, S. Electrochemical epoxidation and carbon-carbon bond cleavage for the preparation of 3-methyl-4-oxo-2-phenyl-4H-1-benzopyran-8-carboxylic acid from 3-methyl-2-phenyl-8-(1-prophenyl)-4H-1-benzopyran-4-one. Bull. Chem. Soc. Jpn. 1985, 58, 2361–2365.
- Inokuchi, T.; Matsumoto, S.; Tsuji, M.; Torii, S. Electrohalogenation of propargyl acetates and amides to form the 1,1-dibromo-2-oxo functionality and a facile synthesis of furaneol. J. Org. Chem. 1992, 57, 5023–5027.
- Inês, M.; Mendonça, A.J.; Esteves, A.P.; Mendonça, D.I.; Medeiros, M.J. Electroepoxidation of natural and synthetic alkenes mediated by sodium bromide. Comptes Rendus Chim. 2009, 12, 841–849.
- Kulangiappar, K.; Ramaprakash, M.; Vasudevan, D.; Raju, T. Electrochemical bromination of cyclic and acyclic enes using biphasic electrolysis. Synth. Commun. 2016, 46, 145–153.
- Yang, C. An impending platinum crisis and its implications for the future of the automobile. Energy Policy 2009, 37, 1805–1808.
- Das, K.K.; Reddy, R.C.; Bagoji, I.B.; Das, S.; Bagali, S.; Mullur, L.; Khodnapur, J.P.; Biradar, M.S. Primary concept of nickel toxicity—An overview. J. Basic Clin. Physiol. Pharmacol. 2019, 30, 141–152.
- Savignan, L.; Faucher, S.; Chéry, P.; Lespes, G. Platinum group elements contamination in soils: Review of the current state. Chemosphere 2021, 271, 129517.
- Sun, X.; Ma, H.; Mei, T.; Fang, P.; Hu, Y. Electrochemical radical formyloxylation-bromination, -chlorination, and -trifluoromethylation of alkenes. Org. Lett. 2019, 21, 3167–3171.
- Bormann, S.; Van Schie, M.M.C.H.; De Almeida, T.P.; Zhang, W.; Stöckl, M.; Ulber, R.; Hollman, F.; Holtmann, D. H2O2 production at low overpotential for electroenzymatic halogenation reactions. ChemSusChem 2019, 12, 4759–4763.
- Wever, R.; Renirie, R.; Hollmann, F. Vanadium chloroperoxidases as versatile biocatalysts. In Vanadium Catalysis; Sudradhar, M., Pombeiro, A.J.L., da Silva, J.A.L., Eds.; Royal Society of Chemistry: London, UK, 2021; Chapter 24; ISBN 978-1-78801-857-9.
- Wu, J.; Abou-Hamdan, H.; Guillot, R.; Kouklovsky, C.; Vincent, G. Electrochemical synthesis of 3a-bromofuranoindolines and 3a-bromopyrroloindolines mediated by MgBr2. Chem. Commun. 2020, 56, 1713–1716.
- Hakamata, H.; Sato, S.; Ueda, H.; Tokuyama, H. AgNTf2-Mediated Allylation with Allylsilanes at C3a-Position of Hexahydropyrroloindoles: Application to Total Syntheses of Amauromine Alkaloids. Org. Lett. 2017, 19, 5308–5311.
- Ramos-Villaseñor, J.M.; Rodríguez-Cárdenas, E.; Díaz, C.E.B.; Frontana-Uribe, B.A. Review—Use of 1,1,1,3,3,3–hexafluoro–2–propanol (HFIP) Co-Solvent Mixtures in Organic Electrosynthesis. J. Electrochem. Soc. 2020, 167, 155509.
- Eberson, L.; Hartshorn, M.P.; Persson, O. 1,1,1,3,3,3-Hexafluoropropan-2-ol as a solvent for the generation of highly persistent radical cations. J. Chem. Soc. Perkin Trans. 2 1995, 1735–1744.
- Eberson, L.; Hartshorn, M.P.; Persson, O. Generation of solutions of highly persistent radical cations by 4-tolylthallium(III) bis(trifluoroacetate) in 1,1,1,3,3,3-hexafluoropropan-2-ol. J. Chem. Soc. Chem. Commun. 1995, 1131–1132.
- Eberson, L.; Persson, O.; Hartshorn, M.P. Detection and Reactions of Radical Cations Generated by Photolysis of Aromatic Compounds with Tetranitromethane in 1,1,1,3,3,3-Hexafluoro-2-propanol at Room Temperature. Angew. Chem. Int. Ed. 1995, 34, 2268–2269.
- Schulz, L.; Waldvogel, S.R. Solvent control in electro-organic synthesis. Synlett 2018, 30, 275–286.
- Röckl, J.L.; Dörr, M.; Waldvogel, S.R. Electrosynthesis 2.0 in 1,1,1,3,3,3-Hexafluoroisopropanol/Amine Mixtures. ChemElectroChem 2020, 7, 3686–3694.
- Ashikari, Y.; Shimizu, A.; Nokami, T.; Yoshida, J. Halogen and Chalcogen Cation Pools Stabilized by DMSO. Versatile Reagents for Alkene Difunctionalization. J. Am. Chem. Soc. 2013, 135, 16070–16073.
- Shimizu, A.; Hayashi, R.; Ashikari, Y.; Nokami, T.; Yoshida, J. Switching the reaction pathways of electrochemically generated β-haloalkoxysulfonium ions—Synthesis of halohydrins and epoxides. Beilstein J. Org. Chem. 2015, 11, 242–248.
- Bityukov, O.V.; Vil’, V.A.; Nikishin, G.I.; Terent’ev, A.O. Alkene, Bromide, and ROH—How To Achieve Selectivity? Electrochemical Synthesis of Bromohydrins and Their Ethers. Adv. Synth. Catal. 2021, 363, 3070–3078.
- Shang, X.; Liu, X.; Sun, Y. Flexible on-site halogenation paired with hydrogenation using halide electrolysis. Green Chem. 2021, 23, 2037–2043.
- Strehl, J.; Abraham, M.L.; Hilt, G. Linear Paired Electrolysis—Realising 200 % Current Efficiency for Stoichiometric Transformations—The Electrochemical Bromination of Alkenes. Angew. Chem. Int. Ed. 2021, 60, 9996–10000.
- Pitzer, L.; Schäfers, F.; Glorius, F. Rapid Assessment of the Reaction-Condition-Based Sensitivity of Chemical Transformations. Angew. Chem. Int. Ed. 2019, 58, 8572–8576.
- Ye, J.H.; Quach, L.; Paulisch, T.; Glorius, F. Visible-light-induced, metal-free carbene insertion into B-H bonds between acylsilanes and pinacolborane. J. Am. Chem. Soc. 2019, 141, 16227–16231.
- Kale, A.P.; Nikolaienko, P.; Smirnova, K.; Rueping, M. Intramolecular Electrochemical Oxybromination of Olefins for the Synthesis of Isoxazolines in Batch and Continuous Flow. Eur. J. Org. Chem. 2021, 2021, 3496–3500.
- He, Y.; Qin, X.; He, X.; Wu, X.; Yin, Z. Practical Synthesis of Halogenated N-Heterocycles via Electrochemical Anodic Oxidation of Unactivated Alkenes. Eur. J. Org. Chem. 2021, 2021, 5831–5834.
- Kim, R.; Ha, J.; Woo, J.; Kim, D.Y. Electrochemical oxidative bromolactonization of unsaturated carboxylic acids with sodium bromide: Synthesis of bromomethylated γ-lactones. Tetrahedron Lett. 2022, 88, 153567.
- Yu, D.; Ji, R.; Sun, Z.; Li, W.; Liu, Z. Electrochemical chlorination and bromination of electron-deficient C-H bonds in quinones, coumarins, quinoxalines and 1,3-diketones. Tetrahedron Lett. 2021, 86, 153514.
- Gombos, L.G.; Werner, L.; Schollmeyer, D.; Martínez-Huitle, C.A.; Waldvogel, S.R. Selective Electrochemical Dibromination of Terpenes and Naturally Derived Olefins. Eur. J. Org. Chem. 2022. accepted.
- Yuan, Y.; Yao, A.; Zheng, Y.; Gao, M.; Zhou, Z.; Qiao, J.; Hu, J.; Ye, B.; Zhao, J.; Wen, H.; et al. Electrochemical Oxidative Clean Halogenation Using HX/NaX with Hydrogen Evolution. iScience 2019, 12, 293–303.
- Seitz, J.; Wirth, T. Electrochemical bromofunctionalization of alkenes in a flow reactor. Org. Biomol. Chem. 2021, 19, 6892–6896.
- Wan, C.; Song, R.; Li, J.H. Electrooxidative 1,2-Bromoesterification of Alkenes with Acids and N-Bromosuccinimide. J. Org. Lett. 2019, 21, 2800–2803.
- Pan, G.; Ouyang, X.; Hu, M.; Xie, Y.; Li, J. Copper-Catalyzed Intermolecular Aminoalkylation of Alkenes with α-Bromoalkyl Esters and Amines toward Pyrrolidin-2-ones. Adv. Synth. Catal. 2017, 359, 2564–2570.
- Klein, M.; Waldvogel, S.R. Counter electrode reactions—Important stumbling blocks on the way to a working electro-organic synthesis. Angew. Chem. Int. Ed. 2022. accepted.
- Zhang, T.; Luo, M.; Li, Y.; Song, R.; Li, J. Electrochemical Alkoxyhalogenation of Alkenes with Organohalides as the Halide Sources via Dehalogenation. Org. Lett. 2020, 22, 7250–7254.
- Dong, X.; Roeckl, J.L.; Waldvogel, S.R.; Morandi, B. Merging shuttle reactions and paired electrolysis for reversible vicinal dihalogenations. Science 2021, 371, 507–514.
More