Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Carmen Berasain | -- | 1430 | 2022-11-14 16:19:23 | | | |
| 2 | Beatrix Zheng | + 3 word(s) | 1433 | 2022-11-16 10:57:18 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Gárate-Rascón, M.; Recalde, M.; Rojo, C.; Fernández-Barrena, M.G.; Ávila, M.A.; Arechederra, M.; Berasain, C. SLU7 in Human Disease. Encyclopedia. Available online: https://encyclopedia.pub/entry/34728 (accessed on 28 July 2026).
Gárate-Rascón M, Recalde M, Rojo C, Fernández-Barrena MG, Ávila MA, Arechederra M, et al. SLU7 in Human Disease. Encyclopedia. Available at: https://encyclopedia.pub/entry/34728. Accessed July 28, 2026.
Gárate-Rascón, María, Miriam Recalde, Carla Rojo, Maite G. Fernández-Barrena, Matías A. Ávila, María Arechederra, Carmen Berasain. "SLU7 in Human Disease" Encyclopedia, https://encyclopedia.pub/entry/34728 (accessed July 28, 2026).
Gárate-Rascón, M., Recalde, M., Rojo, C., Fernández-Barrena, M.G., Ávila, M.A., Arechederra, M., & Berasain, C. (2022, November 15). SLU7 in Human Disease. In Encyclopedia. https://encyclopedia.pub/entry/34728
Gárate-Rascón, María, et al. "SLU7 in Human Disease." Encyclopedia. Web. 15 November, 2022.
Copy Citation
SLU7 (Splicing factor synergistic lethal with U5 snRNA 7) was first identified as a splicing factor necessary for the correct selection of 3′ splice sites, strongly impacting on the diversity of gene transcripts in a cell. More recent studies have uncovered new and non-redundant roles of SLU7 as an integrative hub of different levels of gene expression regulation, including epigenetic DNA remodeling, modulation of transcription and protein stability.
alternative splicing
gene expression
transcription
epigenetics
protein stability
cancer
cirrhosis
hepatocellular identity
targeted therapy
inflammation
1. Introduction
Gene expression is the process by which information encoded in DNA is transformed into functional products: either proteins or non-coding RNA (ncRNA) molecules. Gene expression is finely and dynamically controlled through the tightly coordinated and interconnected activity of multiple factors at different levels [1][2][3]. The first level of regulation is chromatin accessibility, which is determined by epigenetic marks: reversible modifications that, without altering gene sequence, determine chromatin structure and accessibility, leading to the activation or repression of transcription [4]. Transcription of both coding and non-coding genes begins with the recruitment of the RNA polymerase (RNA Pol) and the rest of the transcriptional machinery to the promoter region of the gene [5]. This is followed by the processing of the pre-messenger RNA (pre-mRNA) by splicing, which includes alternative splicing, so that different messenger RNAs (mRNAs) can be generated from the same pre-mRNA, widening the diversity of gene transcripts [6][7]. The mRNA is then directed to the cytoplasm, translated into an amino acid sequence in the ribosome and processed to produce a protein. Once the protein is generated, it can undergo a variety of post-translational modifications that determine its function, localization and half-life (Figure 1). The complexity and interconnection between the different levels of gene expression regulation are necessary to ensure the proper expression of genes in a cell-specific and temporal manner and, thus, guarantee cellular identity, function and viability [2].
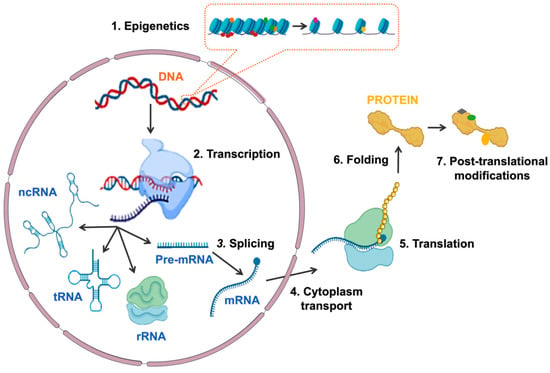
Figure 1. Schematic representation of the different mechanisms involved in regulating gene expression. The sequential regulatory events are: (1) epigenetics modifications at chromatin, (2) transcription of DNA to RNA, (3) splicing of pre-mRNA, (4) mRNA transport to cytoplasm, (5) translation of mRNA to protein, (6) protein folding and (7) protein post-translational modifications. Abbreviations: ncRNA, non-coding RNA; tRNA, transferring RNA; rRNA, ribosomal RNA; Pre-mRNA, pre-messenger RNA; mRNA, messenger RNA.
The splicing factor synergistic lethal with U5 small nuclear RNA, SLU7, was first described in 1992 in yeast as an essential player in the second catalytic reaction of the splicing process [8]. Subsequent works identified its human homologue and characterized its role in the spliceosome [9][10][11][12][13][14], highlighting that SLU7 controls the diversity of gene transcripts in a cell. In addition, more recent findings have identified SLU7 also involved in other levels of gene expression regulation [15][16][17][18][19][20]. Specifically, SLU7 has been implicated in the epigenetic remodeling of DNA, in the modulation of the transcriptional activity and in controlling protein stability. All these findings identify SLU7 as a pleiotropic factor with a holistic function at different levels of gene expression regulation [15][16][17][18][19][20].
2. SLU7 in Human Disease
2.1. SLU7 Expression in Pathological Conditions
Preliminary evidence of changes in SLU7 levels in a pathological condition was in the context of human inflammatory bowel disease [21]. SLU7 was found downregulated in the colonic mucosa of patients with ulcerative colitis independent of the inflammation status of the tissue. Remarkably, in the mucosa of patients with Crohn’s disease, SLU7 was downregulated in inflamed tissues but not in non-inflamed tissues [21]. Therefore, the different expression patterns of SLU7 in these two inflammatory bowel diseases pointed to SLU7 as a potential discriminator between ulcerative colitis and Crohn’s disease [21]. Additionally, genome-wide association studies (GWAS) identified SLU7 single-nucleotide polymorphisms (SNPs) associated with tobacco smoke risk for inflammatory bowel disease (rs41275313) [22].
Evidence demonstrates alterations in SLU7 expression in the liver during the development of liver injury. A significant reduction at the mRNA level [23], that has been recently confirmed at the protein level [19], was observed during the progression of human liver disease from cirrhosis to HCC as well as in mouse models of acute and chronic liver damage [19][23]. This downregulation was associated with the activation of ELK-1 upon binding of AREG to the EGFR and the sequential activation of JNK-1 [23]. Therefore, the induction of AREG expression in the liver in response to inflammatory signals [24][25] would explain the [26] downregulation of SLU7 after liver injury or during the process of hepatocarcinogenesis [15][19][27]. In contrast, hepatic SLU7 was reported to be upregulated in ethanol-fed mice and in patients with alcoholic steatohepatitis, although the underlying mechanism was not described [20]. The induction of SLU7 was also observed in the liver of mice fed a high-fat diet, which could be explained by the presence of insulin resistance, as insulin inhibits SLU7 expression during the physiological cycle of fasting and refeeding [15]. All in all, further studies are needed to clarify the regulation of SLU7 expression in the damaged liver.
Although no link between SLU7 and hematopoiesis has been made so far, SLU7 is located in the region of the long arm of chromosome 5 (from 5q14.1 to 5q35.1) frequently deleted in myelodysplastic syndromes (MDS) [28]; therefore, SLU7 downregulation would be expected in MDS. Additionally, SLU7 has been recently identified as a susceptible locus for systemic lupus erythematosus (rs1895321) [29]. Although these findings highlight the presence of alterations in SLU7 levels during various pathological processes, the implications of these changes in expression are, in most cases, unknown. Therefore, further experiments and studies are needed to find out whether SLU7 changes are implicated in other pathologies, to characterize the mechanisms governing SLU7 expression and to elucidate the role of SLU7 in disease, including liver disease, inflammatory bowel disease and myelodysplastic syndromes. Next, the researchers review the current knowledge of the implications of SLU7 in the context of liver diseases.
2.2. SLU7 in Liver Pathology
Given the role of SLU7 in the maintenance of hepatocellular identity [15], it is reasonable to speculate that the decreased expression of SLU7 observed during the progression of liver disease [19][23] could be involved in the de-differentiation of the hepatocytes and the loss of liver functions observed in patients, which is indicative of their bad prognosis. In fact, the researchers' results have demonstrated that Slu7 haploinsufficiency in mice is enough not only to exacerbate liver dysfunction and potentiate transcriptome rewiring, but also to sensitize the liver to both acute and chronic damage [19]. In fact, all the data concerning the different functions of SLU7 described above suggest that the reduction in SLU7 levels observed in the livers of patients [19][23] contributes to the process of hepatocarcinogenesis. Indeed, it contributes to the reactivation of oncofetal isoforms and proliferation genes [15][30], to the generation of oxidative stress and DNA damage [16], to the induction of genomic instability, both through aneuploidy and mutations [17][31], and to the general DNA hypomethylation [18][32] observed in patients during the process of hepatocarcinogenesis (Figure 2).
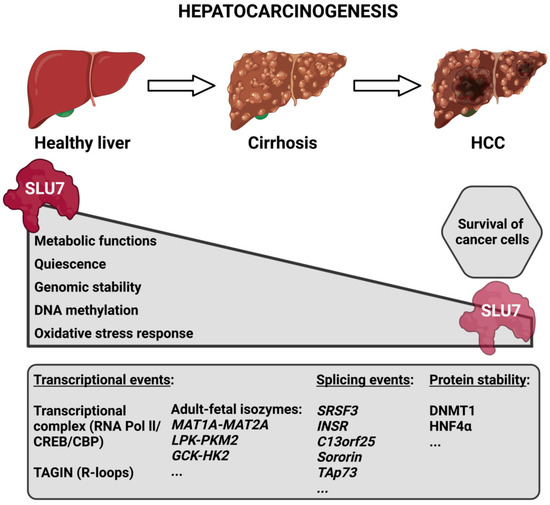
Figure 2. Schematic representation depicting SLU7’s involvement in the process of hepatocarcinogenesis. During the progression of liver disease, the expression of SLU7 decreases in parallel with the alteration of essential cellular functions. However, the residual SLU7 expression in tumoral cells is essential for their survival. The bottom box depicts some SLU7-controlled gene expression events that are altered during liver disease progression. Abbreviations: HCC, hepatocellular carcinoma; TAGIN, transcription-associated genome instability.
Moreover, it is important to note that the researchers' results demonstrate that preventing SLU7 downregulation in damaged mouse liver cells helps to preserve the hepatocellular functions restoring hepatocellular differentiation and, importantly, protects the liver against chronic injury [19]. Therefore, these results reinforce the potential of differentiation therapies such as, for instance, HNF4α1 [33][34] or CBPα [35][36] restitution to treat liver diseases [37], presenting SLU7 as a new target.
2.3. SLU7 in Cancer
Based on the demonstrated functions of SLU7, it is clear that SLU7 is required to preserve cellular homeostasis and genomic integrity. Accordingly, any situation favoring its downregulation could contribute to the process of carcinogenesis, as has been suggested in the liver. However, it is important to highlight that, perhaps due to these central and pleiotropic roles linked to cell proliferation, SLU7 is a non-redundant survival factor for human cancer cells of very different origins and does not affect the viability of normal cells [16]. Thus, in this proliferative context, SLU7 could help to secure cell cycle progression [16] and DNA methylation maintenance [18], the expression of survival oncogenes such as the microRNA cluster miR17-92 [16], and to limit the mitotic stress associated to R-loop accumulation [17].
Therefore, SLU7 could represent a cancer cell’s vulnerability, and the silencing of SLU7 could represent a new therapeutic strategy [16]. In fact, SLU7 knockdown could recapitulate many events that have been proposed separately as anti-cancer strategies, including global DNA demethylation [38][39], induction of R-loop formation [40][41][42][43], DNA damage [40][41], cell cycle arrest [44][45], mitotic stress [40][41], autophagy [46][47] and apoptosis [48][49]. Future studies should help to elucidate whether, as suggested, SLU7 represents a pan-cancer therapeutic target.
References
- Uzman, A. Molecular Biology of the Cell, 4th ed.; Alberts, B., Johnson, A., Lewis, J., Raff, M., Roberts, K., Walter, P., Eds.; Garland Science: New York, NY, USA, 2003.
- Buchberger, E.; Reis, M.; Lu, T.-H.; Posnien, N. Cloudy with a Chance of Insights: Context Dependent Gene Regulation and Impli-cations for Evolutionary Studies. Genes 2019, 10, 492.
- André, K.M.; Sipos, E.H.; Soutourina, J. Mediator Roles Going Beyond Transcription. Trends Genet. 2020, 37, 224–234.
- Baylin, S.B.; Esteller, M.; Rountree, M.R.; Bachman, K.E.; Schuebel, K.; Herman, J.G. Aberrant patterns of DNA methylation, chromatin formation and gene expression in cancer. Hum. Mol. Genet. 2001, 10, 687–692.
- Deutschman, C.S. Transcription. Crit. Care Med. 2005, 33, S400–S403.
- Black, D.L. Protein Diversity from Alternative Splicing A Challenge for Bioinformatics and Post-Genome Biology. Cell 2000, 103, 367–370.
- Kornblihtt, A.R.; Schor, I.E.; Alló, M.; Dujardin, G.; Petrillo, E.; Muñoz, M.J. Alternative splicing: A pivotal step between eukaryotic transcription and translation. Nat. Rev. Mol. Cell. Biol. 2013, 14, 153–165.
- Frank, D.; Patterson, B.; Guthrie, C. Synthetic lethal mutations suggest interactions between U5 small nuclear RNA and four proteins required for the second step of splicing. Mol. Cell. Biol. 1992, 12, 5197–5205.
- Chua, K.; Reed, R. Human step II splicing factor hSlu7 functions in restructuring the spliceosome between the catalytic steps of splicing. Genes Dev. 1999, 13, 841–850.
- Chua, K.; Reed, R. The RNA splicing factor hSlu7 is required for correct 3′ splice-site choice. Nature 1999, 402, 207–210.
- Zhang, X.; Yan, C.; Hang, J.; Finci, L.I.; Lei, J.; Shi, Y. An Atomic Structure of the Human Spliceosome. Cell 2017, 169, 918–929.
- Zhang, X.; Zhan, X.; Yan, C.; Zhang, W.; Liu, D.; Lei, J.; Shi, Y. Structures of the human spliceosomes before and after release of the ligated exon. Cell Res. 2019, 29, 274–285.
- Zhan, X.; Lu, Y.; Zhang, X.; Yan, C.; Shi, Y. Mechanism of exon ligation by human spliceosome. Mol. Cell. 2022, 82, 2769–2778.
- Hegele, A.; Kamburov, A.; Grossmann, A.; Sourlis, C.; Wowro, S.; Weimann, M.; Will, C.L.; Pena, V.; Lührmann, R.; Stelzl, U. Dynamic Protein-Protein Interaction Wiring of the Human Spliceosome. Mol. Cell. 2012, 45, 567–580.
- Elizalde, M.; Urtasun, R.; Azkona, M.; Latasa, M.U.; Goñi, S.; García-Irigoyen, O.; Uriarte, I.; Segura, V.; Collantes, M.; Di Scala, M.; et al. Splicing regulator SLU7 is essential for main-taining liver homeostasis. J. Clin. Investig. 2014, 124, 2909–2920.
- Urtasun, R.; Elizalde, M.; Azkona, M.; Latasa, M.U.; García-Irigoyen, O.; Uriarte, I.; Fernández-Barrena, M.G.; Vicent, S.; Alonso, M.M.; Muntané, J.; et al. Splicing regulator SLU7 preserves survival of hepatocellular carcinoma cells and other solid tumors via oncogenic miR-17-92 cluster expression. Oncogene 2016, 35, 4719–4729.
- Jiménez, M.; Urtasun, R.; Elizalde, M.; Azkona, M.; Latasa, U.M.; Uriarte, I.; Arechederra, M.; Alignani, D.; Barcena-Varela, M.; Álvarez-Sola, G.; et al. Splicing events in the control of genome integrity: Role of SLU7 and truncated SRSF3 proteins. Nucleic Acids Res. 2019, 47, gkz014.
- Recalde, M.; Gárate-Rascón, M.; Elizalde, M.; Azkona, M.; Latasa, M.U.; Bárcena-Varela, M.; Sangro, B.; Fernández-Barrena, M.G.; Ávila, M.A.; Arechederra, M.; et al. The splicing regulator SLU7 is re-quired to preserve DNMT1 protein stability and DNA methylation. Nucleic Acids Res. 2021, 49, gkab649.
- Gárate-Rascón, M.; Recalde, M.; Jimenez, M.; Elizalde, M.; Azkona, M.; Uriarte, I.; Latasa, M.U.; Urtasun, R.; Bilbao, I.; Sangro, B.; et al. SLU7 prevents oxidative stress-mediated HNF4α degradation preserving hepatic differentiation and protecting from liver damage. Hepatology 2021, 74, 2791–2807.
- Wang, J.; Kainrad, N.; Shen, H.; Zhou, Z.; Rote, P.; Zhang, Y.; Nagy, L.E.; Wu, J.; You, M. Hepatic Knockdown of Splicing Regulator Slu7 Ameliorates Inflammation and Attenuates Liver Injury in Ethanol-Fed Mice. Am. J. Pathol. 2018, 188, 1807–1819.
- Häsler, R.; Kerick, M.; Mah, N.; Hultschig, C.; Richter, G.; Bretz, F.; Sina, C.; Lehrach, H.; Nietfeld, W.; Schreiber, S.; et al. Alterations of pre-mRNA splicing in human inflammato-ry bowel disease. Eur. J. Cell. Biol. 2011, 90, 603–611.
- Yadav, P.; Ellinghaus, D.; Rémy, G.; Freitag-Wolf, S.; Cesaro, A.; Degenhardt, F.; Boucher, G.; Delacre, M.; Peyrin-Biroulet, L.; Pichavant, M.; et al. Genetic Factors Interact with Tobacco Smoke to Modify Risk for Inflammatory Bowel Disease in Humans and Mice. Gastroenterology 2017, 153, 550–565.
- Castillo, J.; Goñi, S.; Latasa, M.U.; Perugorría, M.J.; Calvo, A.; Muntané, J.; Bioulac-Sage, P.; Balabaud, C.; Prieto, J.; Ávila, M.A.; et al. Amphiregulin induces the alternative splicing of p73 into its oncogenic isoform DeltaEx2p73 in human hepatocellular tumors. Gastroenterology 2009, 137, 1805–1815.
- Berasain, C.; García-Trevijano, E.R.; Castillo, J.; Erroba, E.; Santamaría, M.; Lee, D.C.; Prieto, J.; Avila, M.A. Novel Role for Amphiregulin in Protec-tion from Liver Injury. J. Biol. Chem. 2005, 280, 19012–19020.
- Berasain, C.; Avila, M.A. Amphiregulin. Semin. Cell Dev. Biol. 2014, 28, 31–41.
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer Genome Landscapes. Science 2013, 339, 1546–1558.
- Berasain, C.; Castillo, J.; Perugorría, M.J.; Prieto, J.; Avila, M.A. Amphiregulin: A new growth factor in hepatocarcinogenesis. Cancer Lett. 2007, 254, 30–41.
- Douet-Guilbert, N.; Soubise, B.; Bernard, D.G.; Troadec, M.-B. Cytogenetic and Genetic Abnormalities with Diagnostic Value in Myelodysplastic Syndromes (MDS): Focus on the Pre-Messenger RNA Splicing Process. Diagnostics 2022, 12, 1658.
- Lessard, C.J.; Adrianto, I.; Ice, J.A.; Wiley, G.B.; Kelly, J.A.; Glenn, S.B.; Adler, A.J.; Li, H.; Rasmussen, A.; Williams, A.H.; et al. Identification of IRF8, TMEM39A, and IKZF3-ZPBP2 as Susceptibility Loci for Systemic Lupus Erythematosus in a Large-Scale Multiracial Replication Study. Am. J. Hum. Genet. 2012, 90, 648–660.
- Chen, X.; Cheung, S.T.; So, S.; Fan, S.T.; Barry, C.; Higgins, J.; Lai, K.M.; Ji, J.; Dudoit, S.; Ng, I.; et al. Gene Expression Patterns in Human Liver Cancers. Mol. Biol. Cell. 2002, 13, 1929–1939.
- Niu, Z.-S.; Niu, X.-J.; Wang, W.-H. Genetic alterations in hepatocellular carcinoma: An update. World. J. Gastroenterol. 2016, 22, 9069–9095.
- Feinberg, A.P.; Ohlsson, R.; Henikoff, S. The epigenetic progenitor origin of human cancer. Nat. Rev. Genet. 2006, 7, 21–33.
- Tafaleng, E.N.; Mukherjee, A.; Bell, A.; Morita, K.; Guzman-Lepe, J.; Haep, N.; Florentino, N.; Diaz-Aragon, R.; Frau, C.; Ostrowska, A.; et al. Hepatocyte Nuclear Factor 4 alpha 2 Messenger RNA Reprograms Liver-Enriched Transcription Factors and Functional Proteins in End-Stage Cirrhotic Human Hepatocytes. Hepatol. Commun. 2021, 5, 1911–1926.
- Yang, T.; Poenisch, M.; Khanal, R.; Hu, Q.; Dai, Z.; Li, R.; Song, G.; Yuan, Q.; Yao, Q.; Shen, X.; et al. Therapeutic HNF4A mRNA attenuates liver fibrosis in a preclinical model. J. Hepatol. 2021, 75, 1420–1433.
- Zhao, X.; Voutila, J.; Ghobrial, S.; Habib, N.A.; Reebye, V. RNA Activation. Adv. Exp. Med. Biol. 2017, 983, 189–194.
- Reebye, V.; Huang, K.-W.; Lin, V.; Jarvis, S.; Cutilas, P.; Dorman, S.; Ciriello, S.; Andrikakou, P.; Voutila, J.; Saetrom, P.; et al. Gene activation of CEBPA using saRNA: Preclinical studies of the first in human saRNA drug candidate for liver cancer. Oncogene 2018, 37, 3216–3228.
- Berasain, C.; Arechederra, M.; Argemí, J.; Fernández-Barrena, M.G.; Avila, M.A. Loss of liver function in chronic liver disease: An identity crisis. J. Hepatol. 2022.
- Stone, M.L.; Chiappinelli, K.B.; Li, H.; Murphy, L.M.; Travers, M.E.; Topper, M.J.; Mathios, D.; Lim, M.; Shih, I.M.; Wang, T.; et al. Reply to Haffner et al.: DNA hypomethylation renders tumors more immunogenic. Proc. Natl. Acad. Sci. USA 2018, 115, E8583-4.
- Topper, M.J.; Vaz, M.; Marrone, K.A.; Brahmer, J.R.; Baylin, S.B. The emerging role of epigenetic therapeutics in immuno-oncology. Nat. Rev. Clin. Oncol. 2020, 17, 75–90.
- Zhang, J.; Dai, Q.; Park, D.; Deng, X. Targeting DNA Replication Stress for Cancer Therapy. Genes 2016, 7, 51.
- Ubhi, T.; Brown, G.W. Exploiting DNA Replication Stress for Cancer Treatment. Cancer Res. 2019, 79, 1730–1739.
- Forment, J.V.; O’Connor, M.J. Targeting the replication stress response in cancer. Pharmacol. Ther. 2018, 188, 155–167.
- Nguyen, H.D.; Zou, L.; Graubert, T.A. Targeting R-loop-associated ATR response in myelodysplastic syndrome. Oncotarget 2019, 10, 2581–2582.
- Schwartz, G.K.; Shah, M.A. Targeting the Cell Cycle: A New Approach to Cancer Therapy. J. Clin. Oncol. 2005, 23, 9408–9421.
- Kumar, G.; Mittal, S.; Parashar, D.; Jadhav, K.; Geethadevi, A.; Cheema, P.S.; Tuli, H.S. Drug Targets in Cellular Processes of Cancer: From Nonclinical to Preclinical Models; Springer: Singapore, 2020; pp. 45–63.
- Janku, F.; McConkey, D.J.; Hong, D.S.; Kurzrock, R. Autophagy as a target for anticancer therapy. Nat. Rev. Clin. Oncol. 2011, 8, 528–539.
- Tschan, M.P.; Simon, H.-U. The role of autophagy in anticancer therapy: Promises and uncertainties. J. Intern. Med. 2010, 268, 410–418.
- Pfeffer, C.M.; Singh, A.T.K. Apoptosis: A Target for Anticancer Therapy. Int. J. Mol. Sci. 2018, 19, 448.
- Pistritto, G.; Trisciuoglio, D.; Ceci, C.; Garufi, A.; D’Orazi, G. Apoptosis as anticancer mechanism: Function and dysfunction of its modulators and targeted therapeutic strategies. Aging 2016, 8, 603–619.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
791
Revisions:
2 times
(View History)
Update Date:
16 Nov 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No