Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Mohamed Chahine | -- | 1783 | 2022-11-14 15:56:42 | | | |
| 2 | Conner Chen | Meta information modification | 1783 | 2022-11-15 09:11:30 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Serres-Bérard, T.D.; Benichou, S.A.; Jauvin, D.; Boutjdir, M.; Puymirat, J.; Chahine, M. Evolution of Chemistries and Pharmaceutical Properties of ASOs. Encyclopedia. Available online: https://encyclopedia.pub/entry/34491 (accessed on 08 August 2026).
Serres-Bérard TD, Benichou SA, Jauvin D, Boutjdir M, Puymirat J, Chahine M. Evolution of Chemistries and Pharmaceutical Properties of ASOs. Encyclopedia. Available at: https://encyclopedia.pub/entry/34491. Accessed August 08, 2026.
Serres-Bérard, Thiéry De, Siham Ait Benichou, Dominic Jauvin, Mohamed Boutjdir, Jack Puymirat, Mohamed Chahine. "Evolution of Chemistries and Pharmaceutical Properties of ASOs" Encyclopedia, https://encyclopedia.pub/entry/34491 (accessed August 08, 2026).
Serres-Bérard, T.D., Benichou, S.A., Jauvin, D., Boutjdir, M., Puymirat, J., & Chahine, M. (2022, November 14). Evolution of Chemistries and Pharmaceutical Properties of ASOs. In Encyclopedia. https://encyclopedia.pub/entry/34491
Serres-Bérard, Thiéry De, et al. "Evolution of Chemistries and Pharmaceutical Properties of ASOs." Encyclopedia. Web. 14 November, 2022.
Copy Citation
Myotonic dystrophy type 1 (DM1) is a dominant genetic disease in which the expansion of long CTG trinucleotides in the 3′ UTR of the myotonic dystrophy protein kinase (DMPK) gene results in toxic RNA gain-of-function and gene mis-splicing affecting mainly the muscles, the heart, and the brain. The CUG-expanded transcripts are a suitable target for the development of antisense oligonucleotide (ASO) therapies. Antisense therapy is an attractive strategy making it possible to target almost every gene by simply modifying the base sequence.
antisense oligonucleotides
myotonic dystrophy type 1
myotonic dystrophy protein kinase
1. Introduction
RNA-based therapies have emerged in the last decades as a promising avenue for regulating various molecular pathways, opening the door for treating diseases that otherwise would be difficult to target with conventional small molecules and recombinant proteins. The development of therapies based on messenger RNAs (mRNAs), RNA aptamers, small interfering RNAs (siRNAs), and antisense oligonucleotides (ASOs) to treat multiple disorders have been the subject of extensive investigations. After several years of slower development, new strategies to enhance the cellular uptake of RNA therapeutics and their resistance to nuclease degradation have boosted their potency and relevance for clinical use. ASOs are short single-stranded sequences of nucleic acid, typically between 15–25 bases long, which are chemically modified to improve their stability and specificity. By hybridizing with their complementary RNA targets, ASOs modulate their processing or their expression, usually by inhibiting their interactions with spliceosomes, ribosomes, and RNA-binding proteins (RBPs) or by promoting their enzymatic degradation. To date, 13 ASO and siRNA drugs have been approved by the U.S. Food and Drug Administration (FDA) for clinical use [1][2], including nusinersen (Spinraza) for treating spinal muscular atrophy (SMA) [3][4], as well as eteplirsen (Exondys 51), golodirsen (Vyondys 53), casimersen (Amondys 45) and viltolarsen (Viltepso) for treating Duchenne muscular dystrophy (DMD) [5][6][7][8].
Myotonic dystrophy type 1 (DM1) is a common muscular dystrophy involving a toxic RNA gain-of-function resulting from the expansion of CTG trinucleotides in the 3′ UTR of the myotonic dystrophy protein kinase (DMPK) gene [9]. The CUG-expanded transcripts adopt stable RNA hairpin conformations and are retained in the nucleus in distinct foci [10]. Splicing factors from the muscleblind-like protein (MBNL) family have a high affinity for CUG repeats and are sequestered by the mutant DMPK mRNAs, impairing their functions, and causing the expression of embryonic splice isoforms in adult tissues [11]. This effect is further aggravated by altered phosphorylation and increased expression of RBPs from the CUGBP Elav-like family (CELF) [12][13][14]. Various mis-splicing events have been associated with a wide range of symptoms manifested by DM1 patients, notably impaired relaxation of muscles (myotonia), muscle weakness, cardiac conduction defects, and cognitive deficits. These symptoms have been recapitulated in different mice models expressing transcripts with expanded CUG repeats, confirming that the toxic RNA gain-of-function is a central mechanism in the disease [15][16][17]. DM1 thus constitutes an ideal candidate for developing pioneer ASO platforms for diseases involving mRNA dysregulation and other microsatellite disorders. In 2014, Ionis Pharmaceuticals initiated the first phase 1/2a DM1 clinical trial based on an antisense strategy (NCT02312011). Ionis reported that subcutaneous administration of multiple escalating doses of the chemically modified IONIS-DMPKRx ASO in adults with DM1 was well tolerated. However, despite inducing minor positive changes in biomarkers and splicing at higher dosages, the ASO concentration measured in muscle biopsies did not reach the therapeutic value of ~10 µg/mg required to support further clinical investigations [18]. Ionis Pharmaceuticals instead announced that it would focus on the development of ligand-conjugated ASOs (LICAs) that bind to specific cell receptors and increase drug concentration and potency in targeted tissues. In recent years, the linkage of ASOs with molecules that facilitate cellular uptake has indeed been an important area of research.
2. Evolution of the Chemistries and Pharmaceutical Properties of ASOs
2.1. First-Generation ASOs
Antisense therapy is an attractive strategy making it possible to target almost every gene by simply modifying the base sequence. However, unmodified nucleic acid oligos are highly sensitive to nucleases and have a limited half-life in vivo. Therefore, various chemical designs have been investigated to improve ASOs’ activity and tolerability (Figure 1). In the first generation of chemically modified ASOs, the phosphate backbone of nucleic acids is substituted by phosphorothioate (PS) bonds where a non-bridging oxygen is replaced by a sulfur atom. It has been known for about 50 years now that this modification makes the oligonucleotides resistant to nuclease degradation, likely because of the larger van der Waals radius of the sulfur atom displacing the metal ions at the active sites of nucleases [19][20][21]. Furthermore, PS-modified ASOs enable the cleavage of the hybridized RNA strand by RNase H [22][23]. As the ASO is still active following target degradation, this mechanism dramatically enhances its inhibitory activity. ASOs synthesized with PS modifications also display an increased affinity for plasma proteins, notably albumin, likely because of interactions between the anionic sulfur and cationic amino acids. ASO binding to serum proteins facilitates cell uptake via the endosomal pathway and slows their excretion in urine following glomerular filtration [24]. However, PS-ASO interactions with paraspeckle proteins may be hepatotoxic [25]. Alternatively, the phosphate backbone may be replaced by N3′-->P5′ phosphoramidate linkages in which the 3′ oxygen of the furanose ring is replaced by a 3′-amino group. Phosphoramidate ASOs are resistant to nucleases and have a high affinity for single-strand RNAs [26][27]. However, they do not activate the RNase H pathway and thus act by steric hindrance to inhibit translation initiation and interactions with RBPs without directly altering the target RNA.
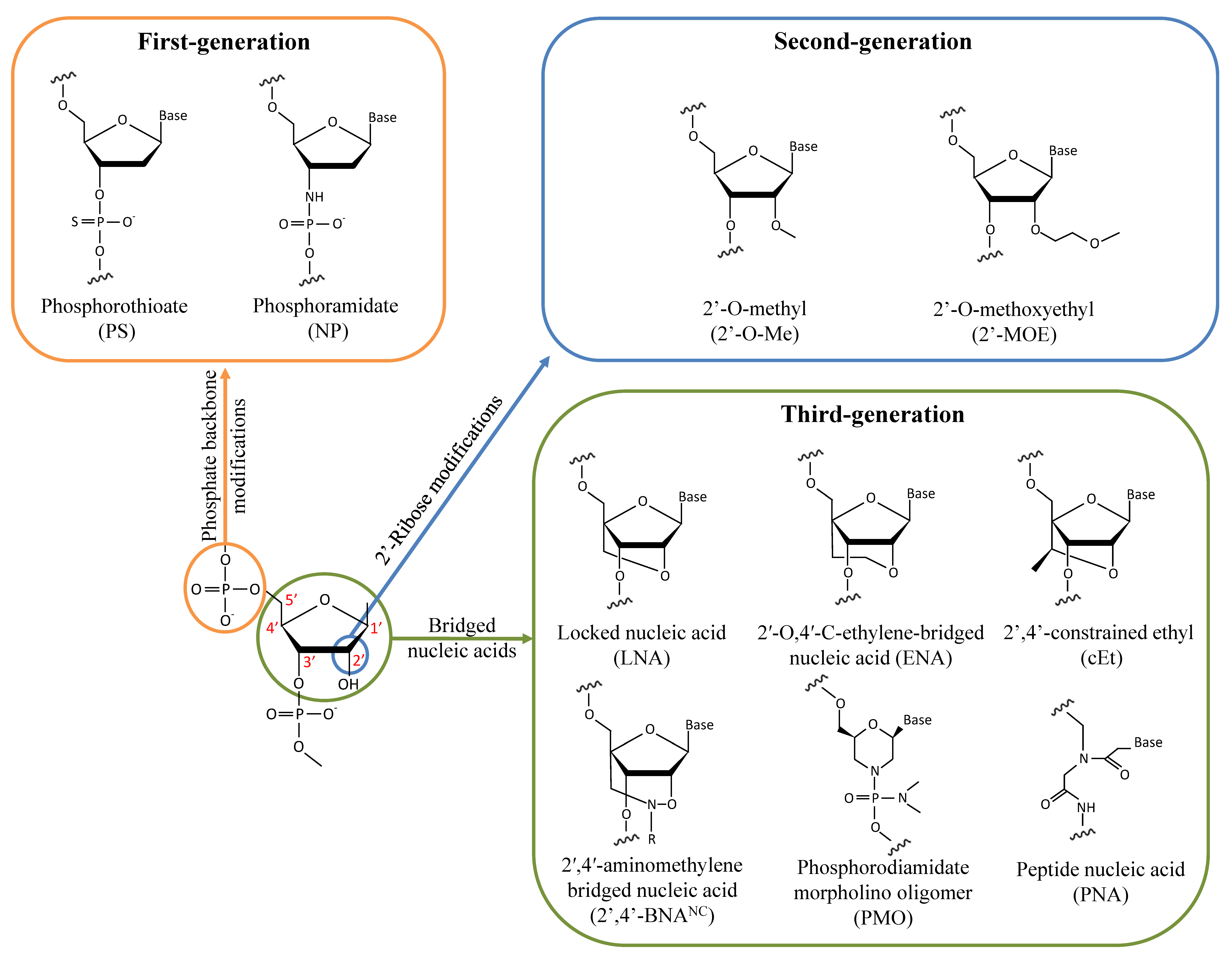
Figure 1. Chemical modifications of the sugar-phosphate backbone of ASOs that have been investigated for treating DM1.
2.2. Second-Generation ASOs
To overcome the limitations of PS modifications, second-generation ASOs were generated by adding alkyl groups to the 2′ hydroxyl of the sugar moiety. Notably, 2′-O-methyl (2′-O-Me) and 2′-O-methoxyethyl (2′-MOE) modifications improve both RNA hybridization and resistance to nucleases while also being less toxic than ASOs modified with PS bonds [28][29][30]. Oligonucleotides with only 2′-alkyl modifications are not recognized by RNase H. Given this, chimeric ASOs called gapmers are often synthesized with a PS or unmodified central gap of deoxyribonucleotide compatible with RNase H activity and are combined with terminal 2′-alkyl modifications to improve their stability. Increasing the RNA hybridization properties of ASOs is especially important to efficiently target RNAs that form secondary structures and that are less accessible to unmodified ASOs, as is the case with CUG-expanded mRNAs [31].
2.3. Third-Generation ASOs
To further improve their pharmacokinetic properties, third-generation ASOs have been designed by modifying the furanose ring. Locked nucleic acids (LNAs) are generated by tethering the 2′ residue of the sugar moiety with the carbon atom at position 4′. The resulting reduced conformational flexibility of the ribose enhances affinity binding to RNAs and may increase the potency 5 to 10-fold with respect to 2′-MOE gapmers [32][33]. Alternatively, the addition of an extra methylene unit in the bridge connecting the 2′ oxygen and the 4′ carbon led to the generation of 2′-O,4′-C-ethylene-bridged nucleic acid (ENA) with better nuclease resistance and RNA affinity properties than the LNA chemistry [34]. LNA ASOs have been reported to be hepatotoxic, limiting their potential for clinical use [33][35][36][37]. A compromise between the LNA and 2′-MOE chemistries has led to the generation of a 2′,4′-constrained ethyl (cEt) modification in which the 2′-O-ethyl substitute is linked to the 4′ position. ASOs with cEt modifications retain a high affinity for RNAs without causing overt toxicity [38][39]. Another LNA analog has been designed by adding an N-O bond to the bridge of the sugar moiety to form a 2′,4′-aminomethylene bridged nucleic acid (2′,4′-BNANC) [40]. The additional nitrogen atom increases ASO target affinity by lowering the repulsion with the negatively charged backbone. The nitrogen atom can also act as a conjugating site to further modify the properties of the ASO by incorporating a hydrogen atom (2′,4′-BNANC[NH]) or a methyl group (2′,4′-BNANC[NME]), which may improve nuclease resistance and target discrimination [40][41]. Substitution of the furanose ring with a morpholine ring combined with phosphorodiamidate modification of the backbone of the oligomer (PMO) is another chemical modification that has been used in a clinical setting. PMO ASOs are neutrally charged, reducing the possibility of interactions with proteins while displaying high resistance to nucleases and enhanced stability [42]. Since PMOs do not activate the RNase H pathway, they are used notably as splice-switching ASOs for treating diseases such as SMA and DMD [43][44]. Another category of nucleotide analog, peptide nucleic acid (PNA), is generated by substituting the phosphate backbone with a pseudopeptide backbone of N-(2-aminoethyl)-glycine linked to the nucleobases by methylene carbonyl [45][46]. PNAs are resistant to nucleases but retain a geometry like that of oligonucleotides and can form stable duplexes with DNA and RNA by Watson–Crick pairing [47]. They have a neutral charge and, thus, bind to RNAs and DNAs with high affinity due to the lack of electrostatic repulsions. As PNAs are randomly folded, combining them with a hydrophilic (R)-diethylene glycol unit (miniPEG) on the γ-backbone of PNAs (MPγPNAs) gives them a right-handed helix conformation, improving both their solubility in water and their affinity for nucleic acids [48]. PNAs do not activate RNase H, and thus, mainly act on their targets by steric hindrance.
2.4. Pharmacokinetics and Cell Distribution of ASOs
Chemical modifications of ASOs are an important factor that determines their pharmacokinetic properties. ASOs are usually systematically administrated by subcutaneous or intravenous injection, absorbed in the plasma, and distributed quickly to tissues, except for the central nervous system (CNS) [49]. Higher ASO concentrations usually accumulate in the liver and the kidneys due to their fenestrated and sinusoidal capillaries, in contrast with cardiac and skeletal muscle tissues, which have a continuous endothelium. The pharmacokinetics of ASOs are characterized by a rapid initial clearance phase from the plasma that occurs over a few hours followed by a slow terminal elimination phase of ASOs absorbed in tissues, which takes place over a few days to several weeks. Negatively charged ASOs, which bind to serum proteins, are more slowly excreted by the kidneys than uncharged ASOs such as PNAs and PMOs. Therefore, these latter usually require conjugates or being administrated at higher doses to be absorbed by tissues at sufficient levels. ASOs are thought to penetrate in myocytes mainly following receptor binding and clathrin-mediated endocytosis, although ASOs’ internalization by clathrin-independent processes may also occur [49][50][51][52]. Only a small portion of ASOs take part in a productive pathway by escaping from endocytic vesicles and shuttling to the nucleus through the nuclear pores, where expanded DMPK mRNAs are localized [52][53]. Most ASOs are retained in endosomes and lysosomes, where they are ultimately degraded and considered non-productive [52]. ASO concentrations in the nucleus in the nanomolar range are required to achieve significant therapeutic effects, with a lower concentration for RNA therapeutics with catalytic activity than for those acting by steric hindrance [54]. Predominant distribution of ASOs with ENA modifications in vesicle-like structures instead of the nucleus in myoblasts has been associated with poor silencing of CUG-expanded DMPK transcripts, in contrast with other chemistries [55].
References
- Roberts, T.C.; Langer, R.; Wood, M.J.A. Advances in Oligonucleotide Drug Delivery. Nat. Rev. Drug Discov. 2020, 19, 673–694.
- Search Results|DrugBank Online. Available online: https://go.drugbank.com/unearth/q?searcher=drugs&query=antisense&approved=1&us=0&ca=0&eu=0&commit=Apply+Filter (accessed on 6 October 2022).
- Finkel, R.S.; Mercuri, E.; Darras, B.T.; Connolly, A.M.; Kuntz, N.L.; Kirschner, J.; Chiriboga, C.A.; Saito, K.; Servais, L.; Tizzano, E.; et al. Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1723–1732.
- Messina, S.; Sframeli, M. New Treatments in Spinal Muscular Atrophy: Positive Results and New Challenges. J. Clin. Med. 2020, 9, 2222.
- Heo, Y.A. Golodirsen: First Approval. Drugs 2020, 80, 329–333.
- Lim, K.R.Q.; Maruyama, R.; Yokota, T. Eteplirsen in the Treatment of Duchenne Muscular Dystrophy. Drug Des. Dev. Ther. 2017, 11, 533–545.
- Charleston, J.S.; Schnell, F.J.; Dworzak, J.; Donoghue, C.; Lewis, S.; Chen, L.; David Young, G.; Milici, A.J.; Voss, J.; Dealwis, U.; et al. Eteplirsen Treatment for Duchenne Muscular Dystrophy: Exon Skipping and Dystrophin Production. Neurology 2018, 90, e2135–e2145.
- Frank, D.E.; Schnell, F.J.; Akana, C.; El-Husayni, S.H.; Desjardins, C.A.; Morgan, J.; Charleston, J.S.; Sardone, V.; Domingos, J.; Dickson, G.; et al. Increased Dystrophin Production with Golodirsen in Patients with Duchenne Muscular Dystrophy. Neurology 2020, 94, e2270–e2282.
- Harley, H.G.; Rundle, S.A.; Reardon, W.; Myring, J.; Crow, S.; Harper, P.S.; Shaw, D.J.; Brook, J.D. Unstable DNA Sequence in Myotonic Dystrophy. Lancet 1992, 339, 1125–1128.
- Mankodi, A.; Urbinati, C.R.; Yuan, Q.P.; Moxley, R.T.; Sansone, V.; Krym, M.; Henderson, D.; Schalling, M.; Swanson, M.S.; Thornton, C.A. Muscleblind Localizes to Nuclear Foci of Aberrant RNA in Myotonic Dystrophy Types 1 and 2. Hum. Mol. Genet. 2001, 10, 2165–2170.
- Yuan, Y.; Compton, S.A.; Sobczak, K.; Stenberg, M.G.; Thornton, C.A.; Griffith, J.D.; Swanson, M.S. Muscleblind-like 1 Interacts with RNA Hairpins in Splicing Target and Pathogenic RNAs. Nucleic Acids Res. 2007, 35, 5474–5486.
- Wei, C.; Stock, L.; Valanejad, L.; Zalewski, Z.A.; Karns, R.; Puymirat, J.; Nelson, D.; Witte, D.; Woodgett, J.; Timchenko, N.A.; et al. Correction of GSK3β at Young Age Prevents Muscle Pathology in Mice with Myotonic Dystrophy Type 1. FASEB J. 2018, 32, 2073–2085.
- Cox, D.C.; Guan, X.; Xia, Z.; Cooper, T.A. Increased Nuclear but Not Cytoplasmic Activities of CELF1 Protein Leads to Muscle Wasting. Hum. Mol. Genet. 2020, 29, 1729.
- Ho, T.H.; Bundman, D.; Armstrong, D.L.; Cooper, T.A. Transgenic Mice Expressing CUG-BP1 Reproduce Splicing Mis-Regulation Observed in Myotonic Dystrophy. Hum. Mol. Genet. 2005, 14, 1539–1547.
- Mankodi, A.; Logigian, E.; Callahan, L.; McClain, C.; White, R.; Henderson, D.; Krym, M.; Thornton, C.A. Myotonic Dystrophy in Transgenic Mice Expressing an Expanded CUG Repeat. Science 2000, 289, 1769–1772.
- Huguet, A.; Medja, F.; Nicole, A.; Vignaud, A.; Guiraud-Dogan, C.; Ferry, A.; Decostre, V.; Hogrel, J.Y.; Metzger, F.; Hoeflich, A.; et al. Molecular, Physiological, and Motor Performance Defects in DMSXL Mice Carrying >1000 CTG Repeats from the Human DM1 Locus. PLoS Genet. 2012, 8, e1003043.
- Mahadevan, M.S.; Yadava, R.S.; Yu, Q.; Balijepalli, S.; Frenzel-Mccardell, C.D.; Bourne, T.D.; Phillips, L.H. Reversible Model of RNA Toxicity and Cardiac Conduction Defects in Myotonic Dystrophy. Nat. Genet. 2006, 38, 1066.
- Ionis Pharmaceuticals—Industry Updates On Drug Development (2018 MDF Annual Conference)|Myotonic Dystrophy Foundation. Available online: https://www.myotonic.org/digital-academy/ionis-pharmaceuticals-industry-updates-drug-development-2018-mdf-annual-conference (accessed on 5 September 2022).
- de Clercq, E.; Eckstein, F.; Merigan, T.C. Interferon Induction Increased through Chemical Modification of a Synthetic Polyribonucleotide. Science 1969, 165, 1137–1139.
- Brautigam, C.A.; Steitz, T.A. Structural Principles for the Inhibition of the 3′-5′ Exonuclease Activity of Escherichia Coli DNA Polymerase I by Phosphorothioates. J. Mol. Biol. 1998, 277, 363–377.
- Stein, C.A.; Subasinghe, C.; Shinozuka, K.; Cohen, J.S. Physicochemical Properties of Phosphorothioate Oligodeoxynucleotides. Nucleic Acids Res. 1988, 16, 3209–3221.
- Walder, R.Y.; Walder, J.A. Role of RNase H in Hybrid-Arrested Translation by Antisense Oligonucleotides. Proc. Natl. Acad. Sci. USA 1988, 85, 5011.
- Kiełpiński, Ł.J.; Funder, E.D.; Schmidt, S.; Hagedorn, P.H. Characterization of Escherichia Coli RNase H Discrimination of DNA Phosphorothioate Stereoisomers. Nucleic Acid Ther. 2021, 31, 383–391.
- Crooke, S.T.; Seth, P.P.; Vickers, T.A.; Liang, X.H. The Interaction of Phosphorothioate-Containing RNA Targeted Drugs with Proteins Is a Critical Determinant of the Therapeutic Effects of These Agents. J. Am. Chem. Soc. 2020, 142, 14754–14771.
- Shen, W.; de Hoyos, C.L.; Migawa, M.T.; Vickers, T.A.; Sun, H.; Low, A.; Bell, T.A.; Rahdar, M.; Mukhopadhyay, S.; Hart, C.E.; et al. Chemical Modification of PS-ASO Therapeutics Reduces Cellular Protein-Binding and Improves the Therapeutic Index. Nat. Biotechnol. 2019, 37, 640–650.
- Faria, M.; Spiller, D.G.; Dubertret, C.; Nelson, J.S.; White, M.R.H.; Scherman, D.; Hélène, C.; Giovannangeli, C. Phosphoramidate Oligonucleotides as Potent Antisense Molecules in Cells and in Vivo. Nat. Biotechnol. 2001, 19, 40–44.
- Gryaznov, S.M.; Lloyd, D.H.; Chen, J.-K.; Schultz, R.G.; Dedionisio, L.A.; Ratmeyert, L.; David Wilsont, W. Oligonucleotide N3′-* P5′ Phosphoramidates. Biochemistry 1995, 92, 5798–5802.
- Sproat, B.S.; Lamond, A.I.; Beijer, B.; Neuner, P.; Ryder, U. Highly Efficient Chemical Synthesis of 2′-O-Methyloligoribonucleotides and Tetrabiotinylated Derivatives; Novel Probes That Are Resistant to Degradation by RNA or DNA Specific Nucleases. Nucleic Acids Res. 1989, 17, 3373.
- Pharmacological Properties of 2-O-Methoxyethyl-Modified Oligonucleotides. Antisense Drug Technol. 2007, 296, 2291–2322.
- Partridge, W.; Xia, S.; Kwoh, T.J.; Bhanot, S.; Geary, R.S.; Baker, B.F. Improvements in the Tolerability Profile of 2′-O-Methoxyethyl Chimeric Antisense Oligonucleotides in Parallel with Advances in Design, Screening, and Other Methods. Nucleic Acid. Ther. 2021, 31, 417.
- Vickers, T.A.; Wyatt, J.R.; Freier, S.M. Effects of RNA Secondary Structure on Cellular Antisense Activity. Nucleic Acids Res. 2000, 28, 1340.
- Vester, B.; Wengel, J. LNA (Locked Nucleic Acid): High-Affinity Targeting of Complementary RNA and DNA. Biochemistry 2004, 43, 13233–13241.
- Swayze, E.E.; Siwkowski, A.M.; Wancewicz, E.; Migawa, M.T.; Wyrzykiewicz, T.K.; Hung, G.; Monia, B.P.; Bennett, C.F. Antisense Oligonucleotides Containing Locked Nucleic Acid Improve Potency but Cause Significant Hepatotoxicity in Animals. Nucleic Acids Res. 2007, 35, 687–700.
- Morita, K.; Hasegawa, C.; Kaneko, M.; Tsutsumi, S.; Sone, J.; Ishikawa, T.; Imanishi, T.; Koizumi, M. 2′-O,4′-C-Ethylene-Bridged Nucleic Acids (ENA): Highly Nuclease-Resistant and Thermodynamically Stable Oligonucleotides for Antisense Drug. Bioorg. Med. Chem. Lett. 2002, 12, 73–76.
- Romero-Palomo, F.; Festag, M.; Lenz, B.; Schadt, S.; Brink, A.; Kipar, A.; Steinhuber, B.; Husser, C.; Koller, E.; Sewing, S.; et al. Safety, Tissue Distribution, and Metabolism of LNA-Containing Antisense Oligonucleotides in Rats. Toxicol. Pathol. 2021, 49, 1174–1192.
- Christou, M.; Wengel, J.; Sokratous, K.; Kyriacou, K.; Nikolaou, G.; Phylactou, L.A.; Mastroyiannopoulos, N.P. Systemic Evaluation of Chimeric LNA/2′-O-Methyl Steric Blockers for Myotonic Dystrophy Type 1 Therapy. Nucleic Acid. Ther. 2020, 30, 80.
- Manning, K.S.; Rao, A.N.; Castro, M.; Cooper, T.A. BNANC Gapmers Revert Splicing and Reduce RNA Foci with Low Toxicity in Myotonic Dystrophy Cells. ACS Chem. Biol. 2017, 12, 2503–2509.
- Seth, P.P.; Siwkowski, A.; Allerson, C.R.; Vasquez, G.; Lee, S.; Prakash, T.P.; Kinberger, G.; Migawa, M.T.; Gaus, H.; Bhat, B.; et al. Design, Synthesis and Evaluation of Constrained Methoxyethyl (CMOE) and Constrained Ethyl (CEt) Nucleoside Analogs. Nucleic Acids Symp. Ser. 2008, 52, 553–554.
- Pallan, P.S.; Allerson, C.R.; Berdeja, A.; Seth, P.P.; Swayze, E.E.; Prakash, T.P.; Egli, M. Structure and Nuclease Resistance of 2′,4′-Constrained 2′-O-Methoxyethyl (CMOE) and 2′-O-Ethyl (CEt) Modified DNAs. Chem. Commun. 2012, 48, 8195.
- Rahman, S.M.A.; Seki, S.; Obika, S.; Yoshikawa, H.; Miyashita, K.; Imanishi, T. Design, Synthesis, and Properties of 2′,4′-BNA NC: A Bridged Nucleic Acid Analogue. J. Am. Chem. Soc. 2008, 130, 14.
- Miyashita, K.; Rahman, S.M.A.; Seki, S.; Obika, S.; Imanishi, T. N-Methyl Substituted 2′,4′- BNANC: A Highly Nuclease-Resistant Nucleic Acid Analogue with High-Affinity RNA Selective Hybridization. Chem. Commun. 2007, 36, 3765–3767.
- Summerton, J.; Weller, D. Morpholino Antisense Oligomers: Design, Preparation, and Properties. Antisense Nucleic Acid. Drug. Dev. 1997, 7, 187–195.
- Son, H.W.; Yokota, T. Recent Advances and Clinical Applications of Exon Inclusion for Spinal Muscular Atrophy. Methods Mol. Biol. 2018, 1828, 57–68.
- Tsoumpra, M.K.; Fukumoto, S.; Matsumoto, T.; Takeda, S.; Wood, M.J.A.; Aoki, Y. Peptide-Conjugate Antisense Based Splice-Correction for Duchenne Muscular Dystrophy and Other Neuromuscular Diseases. EBioMedicine 2019, 45, 630–645.
- Nielsen, P.E.; Egholm, M.; Berg, R.H.; Buchardt, O. Sequence-Selective Recognition of DNA by Strand Displacement with a Thymine-Substituted Polyamide. Science 1991, 254, 1497–1500.
- Nielsen, P.E. PNA Technology. Mol. Biotechnol. 2004, 26, 233–248.
- Egholm, M.; Buchardt, O.; Christensen, L.; Behrens, C.; Freier, S.M.; Driver, D.A.; Berg, R.H.; Kim, S.K.; Norden, B.; Nielsen, P.E. PNA Hybridizes to Complementary Oligonucleotides Obeying the Watson-Crick Hydrogen-Bonding Rules. Nature 1993, 365, 566–568.
- Sahu, B.; Sacui, I.; Rapireddy, S.; Zanotti, K.J.; Bahal, R.; Armitage, B.A.; Ly, D.H. Synthesis and Characterization of Conformationally-Preorganized, MiniPEG-Containing ΓPNAs with Superior Hybridization Properties and Water Solubility. J. Org. Chem. 2011, 76, 5614.
- Geary, R.S.; Norris, D.; Yu, R.; Bennett, C.F. Pharmacokinetics, Biodistribution and Cell Uptake of Antisense Oligonucleotides. Adv. Drug. Deliv. Rev. 2015, 87, 46–51.
- Koller, E.; Vincent, T.M.; Chappell, A.; De, S.; Manoharan, M.; Bennett, C.F. Mechanisms of Single-Stranded Phosphorothioate Modified Antisense Oligonucleotide Accumulation in Hepatocytes. Nucleic Acids Res. 2011, 39, 4795–4807.
- Miller, C.M.; Donner, A.J.; Blank, E.E.; Egger, A.W.; Kellar, B.M.; Østergaard, M.E.; Seth, P.P.; Harris, E.N. Stabilin-1 and Stabilin-2 Are Specific Receptors for the Cellular Internalization of Phosphorothioate-Modified Antisense Oligonucleotides (ASOs) in the Liver. Nucleic Acids Res. 2016, 44, 2782–2794.
- González-Barriga, A.; Nillessen, B.; Kranzen, J.; van Kessel, I.D.G.; Croes, H.J.E.; Aguilera, B.; de Visser, P.C.; Datson, N.A.; Mulders, S.A.M.; van Deutekom, J.C.T.; et al. Intracellular Distribution and Nuclear Activity of Antisense Oligonucleotides After Unassisted Uptake in Myoblasts and Differentiated Myotubes In Vitro. Nucleic Acid. Ther. 2017, 27, 144.
- Leonetti, J.P.; Mechti, N.; Degols, G.; Gagnor, C.; Lebleu, B. Intracellular Distribution of Microinjected Antisense Oligonucleotides. Proc. Natl. Acad. Sci. USA 1991, 88, 2702.
- van der Bent, M.L.; da Silva Filho, O.P.; Willemse, M.; Hällbrink, M.; Wansink, D.G.; Brock, R. The Nuclear Concentration Required for Antisense Oligonucleotide Activity in Myotonic Dystrophy Cells. FASEB J. 2019, 33, 11314–11325.
- González-Barriga, A.; Mulders, S.A.M.; van de Giessen, J.; Hooijer, J.D.; Bijl, S.; van Kessel, I.D.G.; van Beers, J.; van Deutekom, J.C.T.; Fransen, J.A.M.; Wieringa, B.; et al. Design and Analysis of Effects of Triplet Repeat Oligonucleotides in Cell Models for Myotonic Dystrophy. Mol. Ther. Nucleic Acids 2013, 2, e81.
More
Information
Subjects:
Medicine, General & Internal
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
959
Revisions:
2 times
(View History)
Update Date:
15 Nov 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No