Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Mohamed Chahine and Version 2 by Conner Chen.
Myotonic dystrophy type 1 (DM1) is a dominant genetic disease in which the expansion of long CTG trinucleotides in the 3′ UTR of the myotonic dystrophy protein kinase (DMPK) gene results in toxic RNA gain-of-function and gene mis-splicing affecting mainly the muscles, the heart, and the brain. The CUG-expanded transcripts are a suitable target for the development of antisense oligonucleotide (ASO) therapies. Antisense therapy is an attractive strategy making it possible to target almost every gene by simply modifying the base sequence.
- antisense oligonucleotides
- myotonic dystrophy type 1
- myotonic dystrophy protein kinase
1. Introduction
RNA-based therapies have emerged in the last decades as a promising avenue for regulating various molecular pathways, opening the door for treating diseases that otherwise would be difficult to target with conventional small molecules and recombinant proteins. The development of therapies based on messenger RNAs (mRNAs), RNA aptamers, small interfering RNAs (siRNAs), and antisense oligonucleotides (ASOs) to treat multiple disorders have been the subject of extensive investigations. After several years of slower development, new strategies to enhance the cellular uptake of RNA therapeutics and their resistance to nuclease degradation have boosted their potency and relevance for clinical use. ASOs are short single-stranded sequences of nucleic acid, typically between 15–25 bases long, which are chemically modified to improve their stability and specificity. By hybridizing with their complementary RNA targets, ASOs modulate their processing or their expression, usually by inhibiting their interactions with spliceosomes, ribosomes, and RNA-binding proteins (RBPs) or by promoting their enzymatic degradation. To date, 13 ASO and siRNA drugs have been approved by the U.S. Food and Drug Administration (FDA) for clinical use [1][2][1,2], including nusinersen (Spinraza) for treating spinal muscular atrophy (SMA) [3][4][3,4], as well as eteplirsen (Exondys 51), golodirsen (Vyondys 53), casimersen (Amondys 45) and viltolarsen (Viltepso) for treating Duchenne muscular dystrophy (DMD) [5][6][7][8][5,6,7,8].
Myotonic dystrophy type 1 (DM1) is a common muscular dystrophy involving a toxic RNA gain-of-function resulting from the expansion of CTG trinucleotides in the 3′ UTR of the myotonic dystrophy protein kinase (DMPK) gene [9]. The CUG-expanded transcripts adopt stable RNA hairpin conformations and are retained in the nucleus in distinct foci [10]. Splicing factors from the muscleblind-like protein (MBNL) family have a high affinity for CUG repeats and are sequestered by the mutant DMPK mRNAs, impairing their functions, and causing the expression of embryonic splice isoforms in adult tissues [11]. This effect is further aggravated by altered phosphorylation and increased expression of RBPs from the CUGBP Elav-like family (CELF) [12][13][14][12,13,14]. Various mis-splicing events have been associated with a wide range of symptoms manifested by DM1 patients, notably impaired relaxation of muscles (myotonia), muscle weakness, cardiac conduction defects, and cognitive deficits. These symptoms have been recapitulated in different mice models expressing transcripts with expanded CUG repeats, confirming that the toxic RNA gain-of-function is a central mechanism in the disease [15][16][17][15,16,17]. DM1 thus constitutes an ideal candidate for developing pioneer ASO platforms for diseases involving mRNA dysregulation and other microsatellite disorders. In 2014, Ionis Pharmaceuticals initiated the first phase 1/2a DM1 clinical trial based on an antisense strategy (NCT02312011). Ionis reported that subcutaneous administration of multiple escalating doses of the chemically modified IONIS-DMPKRx ASO in adults with DM1 was well tolerated. However, despite inducing minor positive changes in biomarkers and splicing at higher dosages, the ASO concentration measured in muscle biopsies did not reach the therapeutic value of ~10 µg/mg required to support further clinical investigations [18]. Ionis Pharmaceuticals instead announced that it would focus on the development of ligand-conjugated ASOs (LICAs) that bind to specific cell receptors and increase drug concentration and potency in targeted tissues. In recent years, the linkage of ASOs with molecules that facilitate cellular uptake has indeed been an important area of research.
2. Evolution of the Chemistries and Pharmaceutical Properties of ASOs
2.1. First-Generation ASOs
Antisense therapy is an attractive strategy making it possible to target almost every gene by simply modifying the base sequence. However, unmodified nucleic acid oligos are highly sensitive to nucleases and have a limited half-life in vivo. Therefore, various chemical designs have been investigated to improve ASOs’ activity and tolerability (Figure 1). In the first generation of chemically modified ASOs, the phosphate backbone of nucleic acids is substituted by phosphorothioate (PS) bonds where a non-bridging oxygen is replaced by a sulfur atom. It has been known for about 50 years now that this modification makes the oligonucleotides resistant to nuclease degradation, likely because of the larger van der Waals radius of the sulfur atom displacing the metal ions at the active sites of nucleases [19][20][21][19,20,21]. Furthermore, PS-modified ASOs enable the cleavage of the hybridized RNA strand by RNase H [22][23][22,23]. As the ASO is still active following target degradation, this mechanism dramatically enhances its inhibitory activity. ASOs synthesized with PS modifications also display an increased affinity for plasma proteins, notably albumin, likely because of interactions between the anionic sulfur and cationic amino acids. ASO binding to serum proteins facilitates cell uptake via the endosomal pathway and slows their excretion in urine following glomerular filtration [24]. However, PS-ASO interactions with paraspeckle proteins may be hepatotoxic [25]. Alternatively, the phosphate backbone may be replaced by N3′-->P5′ phosphoramidate linkages in which the 3′ oxygen of the furanose ring is replaced by a 3′-amino group. Phosphoramidate ASOs are resistant to nucleases and have a high affinity for single-strand RNAs [26][27][26,27]. However, they do not activate the RNase H pathway and thus act by steric hindrance to inhibit translation initiation and interactions with RBPs without directly altering the target RNA.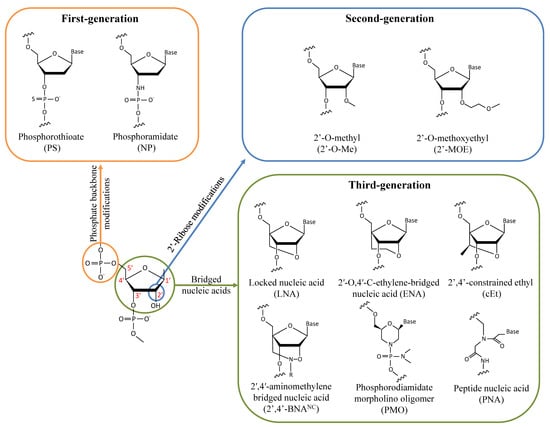
Figure 1.
Chemical modifications of the sugar-phosphate backbone of ASOs that have been investigated for treating DM1.