1. Atherosclerotic Plaque Formation and Development of Inflammation
The stressors discussed above subject the endothelial cells (EC) to injury, so they undergo alterations in their adhesiveness, becoming ‘sticky,’ leading to monocyte and T cell attachment. Dysfunctional ECs also exhibit growth-stimulatory characteristics, enabling the entry of LDL molecules and monocytes into the intimal layer, triggering the formation of a fatty streaks, the first step in the long process of plaque formation [1][2].
The LDL particles that enter the subintimal layer get oxidized. The oxidized LDL (ox-LDL) induces the release of chemical mediators by the cells in its vicinity and promotes the accumulation of macrophages that phagocytose the Ox-LDL. This process initiates the plaque formation and recruits more inflammatory cells, beginning the process of chronic inflammation in the arterial wall [3][4].
2. Vascular Smooth Muscle Cells (VSMCs)
Within the growing stable plaque, an interaction between the T cells and macrophages secrete a wide array of chemokines and growth factors that target circulating monocytes, local endothelial cells, and smooth muscle cells. This proliferation can lead to lesion expansion. As discussed above, the stability of the plaque is derived from the presence of a thick fibrous cap produced in part by the VSMCs. Activated VSMCs switch their phenotype from contractile to synthetic and synthesize fibrotic proteins such as collagen
[5]. Typically, TGF-β is a potent activator of collagen synthesis by VSMCs; however, T-cells secrete INF-γ, which inhibits collagen synthesis, especially types I and III, that are majorly found in extracellular matrices of arteries
[6][7]. It has been shown that INF-γ inhibit collagen production by VSMCs even in the presence of TGF-β
[7]. The source of INF-γ is T-cells, the presence of which in atherosclerotic lesions helps to tie the link between adaptive immune response to the stability of a plaque.
VSMCs also express scavenger receptors such as LOX-1 and CD-36 that internalize ox-LDL, generating foam cells out of VSMCs, as they would with macrophages. This generation of foam cells from VSMCs is most likely due to VSMCs’ phenotypic change to resemble monocytes and mesenchymal stem cells. Endothelial and VSMCs, under the conditions of acute inflammation, start to produce pro-inflammatory molecules such as TNF-α, IL-1β, and MCP-1, among many others, which attract neutrophils to the inflammatory site. Neutrophils, in turn, act on these cells to increase their pro-inflammatory effects, thus producing a snowball effect that prolongs the inflammatory response and intimal hyperplasia
[8]. Studies have shown that a knockout of the transcription factor, Klf-4, which possibly mediates the phenotypic change in VSMCs, results in a reduction of VSMC-derived macrophage-like cells, a smaller lesion size, and increased fibrous cap thickness. These changes consequently increase plaque stability
[9].
3. Macrophages, T Cells, Platelets
Macrophages, classically have been categorized into M1, involved in pro-inflammatory pathways, and M2, participating in anti-inflammatory events. As noted above, monocytes recruited to the site of plaque formation are activated by toll-like receptors (TLRs), and INF-γ present in the micro-environment of a growing plaque to M1 subtype
[10][11][12]. These M1 type macrophages go on to secrete other pro-inflammatory cytokines such as IL-1b, IL-6, TNF, IL-12, and IL-23 along with molecules such as reactive oxygen and nitrogen species that sustain the ongoing inflammation
[10]. M1 type macrophage also recruit Th1 and natural killer cells to the growing plaque further exacerbating the injury due to uncontrolled inflammation
[13].
While historically, atherosclerotic plaques were characterized mainly by M1 type macrophages, recent studies have brought to fore other subtypes of macrophages, such as M4, Mox, M(Hb), Mhem. These subtypes vary in their local milieu and therefore their activation, their phenotypic markers, and in their participation or role in the pathogenesis as well as the characteristics of an atherosclerotic plaque. Recent studies with mouse models and carotid plaques have shown the presence of M2 subtype in advanced lesions
[14]. However, since the cause of their presence could not be narrowed down, and since macrophage phenotypic switching based on their micro-environment is well-known, such representation is challenging and studies are still ongoing to demonstrate the presence of distinct types of macrophages and their role in plaque stability and severity
[10].
The reduction in collagen levels in a plaque is partly due to decreased production mediated by T cells and increased levels of breakdown by macrophages, neutrophils, endothelial cells, and SMCs. Studies have shown that macrophages secrete several matrix metalloproteinases (MMPs), such as MMP-1, MMP-2, MMP-8, MMP-9, and MMP-13, which are structurally identical to enzymes that degrade fibrillar collagen types I and III, proteoglycans, and elastin. Such MMPs also activate platelet aggregation and adhesion. Ox-LDL increases MMP-14 expression in endothelial cells, which in turn activates MMP-2, a potent gelatinase that acts on collagen IV, a constituent of the basement membrane. T-cells can bring such secretions by macrophages through the CD40 ligand (CD40L). The finding that the lack of MMPs resulted in increased and better-organized collagen in such plaques proved the role of MMPs in plaque stability. Elastases such as cathepsins S and K, and neutrophil elastase, have been found in plaques, which lower plaque stability possibly by degrading the extra-cellular matrix
[7]. CD40L by T cells not only increases the secretion of MMPs but has also been attributed to the expression of tissue factor (TF) expression by macrophages. TF is the major activator of thrombosis when the cells encounter coagulation factors in blood. CD40L are also derived from platelets, which can cause its aggregation when the local environment is conducive. This can lead to local small arteries that feed the plaque to rupture leading to an intraplaque hemorrhage. This loop can continue whereby the exposed CD40L can activate TF in the local environment, leading to the growth of the thrombus, which in turn can cause further inflammation. While rupture of a plaque is the climax of the negative cardiac events, superficial erosion of the endothelial cells lining the plaque contributes to the weakening of its cap
[7].
Not all small-scale changes that occur regularly within a plaque that result in mural thrombus formation lead to negative cardiac events, as seen in the thrombus formed within the vasculature of patients that did not die of heart conditions. These mural thrombi have platelets as its participants and platelet derived growth factor (PDGF) thus secreted might have led to the fibrosis of the plaque conferring it with stability. This ‘healing’ of the minor plaque ruptures lead to the ‘expansive remodeling’ of the artery rather than the ‘constrictive remodeling’ observed in an immature growing inflamed plaque
[7].
4. Apoptosis
TNF family of cytokines from platelets has a pronounced effect on the apoptosis of VSMCs. In addition, chemokines such as IL-1beta, TNF-alpha, and IFN-gamma by activated macrophages and T cells in the immediate environment, induce apoptosis in VSMC and block collagen production
[15]. Myeloperoxidase produces hypochlorous acid in the event of increased oxidative stress, which induces apoptosis in EC. Increased caspase-3 and DNA ladders in ECs support the theory that oxidative stress inherent in inflammation-induced plaque formation leads to a cyclic deterioration of plaque stability
[7].
Loss of collagen through apoptosis of VSMCs, increased MMPs, and the accumulation of necrotic debris by apoptotic macrophages, all cumulatively result in an unstable plaque, increasing the chance for rupture. Risk of plaque rupture also increases with superficial erosion of endothelial monolayer, the risk of which is increased with apoptosis of endothelial cells due to inflammation
[7]. Apoptosis is beneficial in clearing the cells that help eliminate oxidized elements in the initial lesion, but the same processes, if left unchecked, are detrimental during the later stages. Removing the injurious agent or resolving the inflammation could reverse the progression of the lesion from a fatty streak to an unstable plaque.
5. Resolution of Inflammation
Resolution of inflammation has been established as an active process that begins with and is characterized by a reduction in neutrophil recruitment and an increase in efferocytosis, a non-phlogistic clearance of cellular debris by macrophages
[16].
As with the active inflammation process, its resolution involves a myriad of chemical modulators from a variety of cell populations through complex chemical pathways that are interconnected. Resolution of an ongoing inflammation is kicked off by the “class switching” of prostaglandins and leukotrienes to lipoxins, which are also derived from arachidonic acid
[17].
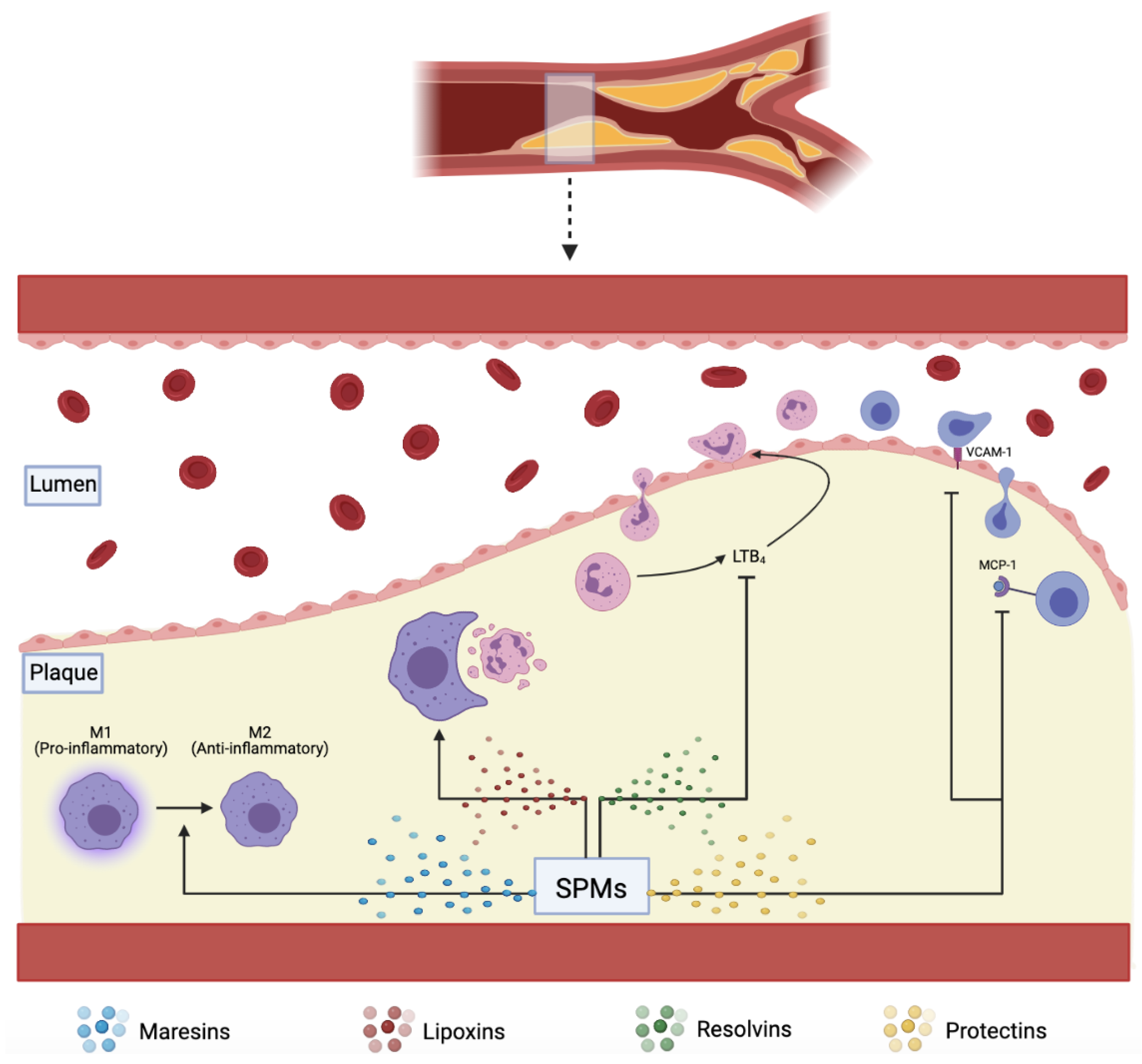
The resolution of acute inflammation was studied by analyzing the exudate from inflamed tissues and was found to be mediated by molecules derived from essential fatty acids, Eicosapentaenoic acid, and Docosahexaenoic acid (DHA). These molecules are termed SPMs and are classified into further subdivisions—resolvins (E and D series) described in more details below, protectins, lipoxins, and maresins. E-series resolvins are generated from EPA, lipoxins from arachidonic acid, while the rest, D-series resolvins, protectins, and Maresins, are derived from DHA
[9][18]. These SPMs assist in reducing inflammation via several mechanisms, including increasing efferocytosis, as shown in (
Figure 1)
[19].
Figure 1. SPMs including maresins, lipoxins, resolvins, and protectins, assist in plaque resolution through various pathways: converting pro-inflammatory M1 to anti-inflammatory M2 macrophages, increasing effective efferocytosis, downregulating pro-inflammatory LTB4, VCAM-1, and MCP-1.
6. Efferocytosis
The process in which apoptotic cells are cleared is called efferocytosis. While mainly managed by macrophages, vascular smooth muscle cells and neighboring cells may also have efferocytotic roles. As discussed above, apoptosis is a significant phenomenon that defines the progression of a plaque: EC, VSMC, and foamy macrophages all undergo apoptosis fueled by the growing plaque’s chemokine environment. Tabas et al. have concluded that clearance of such apoptotic cells is the real issue within an atherosclerotic plaque than apoptosis itself
[2]. Martinet et al. have shown that efferocytosis is reduced by approximately 20-fold in a plaque relative to normal
[20][21]. Interestingly, macrophages that become foam cells do not unload the engulfed ox-LDL to HDLs due to their defective efferocytosis, suppressing reverse cholesterol pathways’ normal functioning. Defective efferocytosis also triggers macrophages to secrete pro-inflammatory signals such as TGF-β or IL-10. When efferocytosis is not complete, macrophages undergo cell membrane lysis spilling out necrotic chemicals such as proteases, thrombogenic tissue factors, and angiogenesis-promoting cytokines, creating more pro-inflammatory pathways (
Figure 1). It is also thought that phenotype switching of macrophages from anti-inflammatory M2 to pro-inflammatory M1 has a role in diminishing the efficiency of efferocytosis
[22].
Several classes of cellular molecules highly regulate efferocytosis: “find me” ligands that recruit phagocytes to the site of apoptosis, bridging molecules that link phagocytes to their targets, and “eat me” ligands on the apoptotic cell surface. These “eat me” ligands on cell surfaces bind and activate engulfment receptors on phagocytes. A counter molecule class called “don’t eat me” ligand is present on viable cells but is downregulated in apoptotic cells. Studies have shown that ox-LDL in plaques induces auto-antibody generation within macrophages and other phagocytes. These autoantibodies mask the cell-surface “eat me” ligands on the dying cell. Ox-LDL also seems to act as a competitive inhibitor of scavenger receptors, making them less efficient in clearing apoptotic cells. Calreticulin (Calr) is one of the key “eat me” ligands that binds to LDL Receptor-Related Protein (LRP1) on phagocytic cells and induces engulfment. Carriers of a risk allele at chromosome 9p21 are shown to express less Calr due to an inherited defect in TGF-β signaling, resulting in a more extensive lesion formation. Plaques of such mice models (that lack one of the 9p21 candidate genes) have been shown to exhibit plaque destabilizing features. VSMCs deficient in Calr have been shown to resist phagocytosis in vitro, induce pro-inflammatory foam-cell phenotype on cocultured macrophages, and suppress reverse cholesterol transport. It was shown that an exogenous introduction of Calr reversed these effects in-vitro
[22].
As mentioned above, the “don’t eat me” ligands maintain the balance of efferocytosis and protect healthy cells from being phagocytosed. CD47 is one such ligand on healthy cells that interacts with the alpha receptor on phagocytes, shutting off the efferocytotic pathways within the phagocytes. TNF-α weakens the downregulation of CD47 in atherosclerotic plaque cells and renders them resistant to efferocytosis. CD47 blocking antibodies have shown to have beneficial effects in mouse models by preventing atherosclerotic progression, regressing the necrotic core, and preventing the plaque from rupturing
[22].
A few other molecules have been implicated in the failure of efferocytosis in atherosclerosis, including Milk fat globule epidermal growth factor 8 (Mfge8) and Mer receptor tyrosine kinase (Mertk). Mfge8 is a bridging molecule between αvβ3 integrin on the macrophages and externalized phosphatidyl serine on the apoptotic body
[23]. This molecule seems to be expressed less in atherosclerotic plaque. In mouse models that were created with both LDLR−/−, and transplanted Mfge8−/− bone marrow had advanced atherosclerosis with larger necrotic core and systemic inflammation. It is thought that Mfge8 might also have a role in reverse cholesterol transport by binding transglutaminase 2
[22].
Mouse models with both LDLR−/−, and transplanted Mertk−/− bone marrow showed similar plaque properties to those with absent Mfge8 and LDLR. Furthermore, a mouse with a defective kinase form of Mertk resulted in more plaque necrosis than those found in ApoE−/−. Metalloproteinases, generally found in abundance in pro-inflammatory settings, cleave Mertk into a soluble inactive form. This inactive molecule provides a decoy receptor to Growth Arrest Specific 6 and leads to competitive inhibition of efferocytosis
[22].
Weissman and colleagues have found that cancer cells upregulate “don’t eat me” ligands to evade phagocytosis. Antibodies and decoy molecules that inhibit such processes and restore normal phagocytosis have been developed and are under study. If such treatments prove effective, they will have immense potential in treating atherosclerosis. Similarly, antibodies to TNF-α have been shown to reduce the expression of CD47. Anti-TNF-α antibodies are used in patients with rheumatological conditions. Such patients appear to be protected from myocardial infarction or adverse cardiovascular effects. Mouse treated with a combination of anti-CD47 and anti-TNF-α antibodies showed a better reduction in atherosclerosis than anti-CD47 alone. Given that there is a genetic susceptibility in reducing the efficiency of efferocytosis through the example of Calr expression, a genotype-driven therapy will most benefit such individuals
[22].
7. Resolvins
7.1. Resolvin D Series (RvD)
RvD1 binds to two receptors, ChemR23 and BLT1, and increases macrophage phagocytosis and PMN apoptosis, respectively. RvD1 also upregulates anti-inflammatory IL-10 and downregulates pro-inflammatory LTB4
[8]. Overexpression of the enzyme 15-Lipoxygenase (15-LOX) reduced atherosclerotic plaques in rabbits
[16]. Lack of resolution of inflammation and the increased ratio of LTB4 to RvD1 have been implicated as the real culprit behind what starts as a host-beneficial process to life-threatening events
[24][25]. Since SPMs act in their local environments, the microclimate of the area of tissue necrosis is vital in determining their viability and action; thus, a 5-LOX closer to the cell periphery can interact with 12/15 LOX to produce RvD1 from DHA. Furthermore, RvD1 prevents the nuclear location of 5-LOX, increasing the production of pro-resolving LXA4 and reducing the production of LTB4 (
Figure 2)
[26].
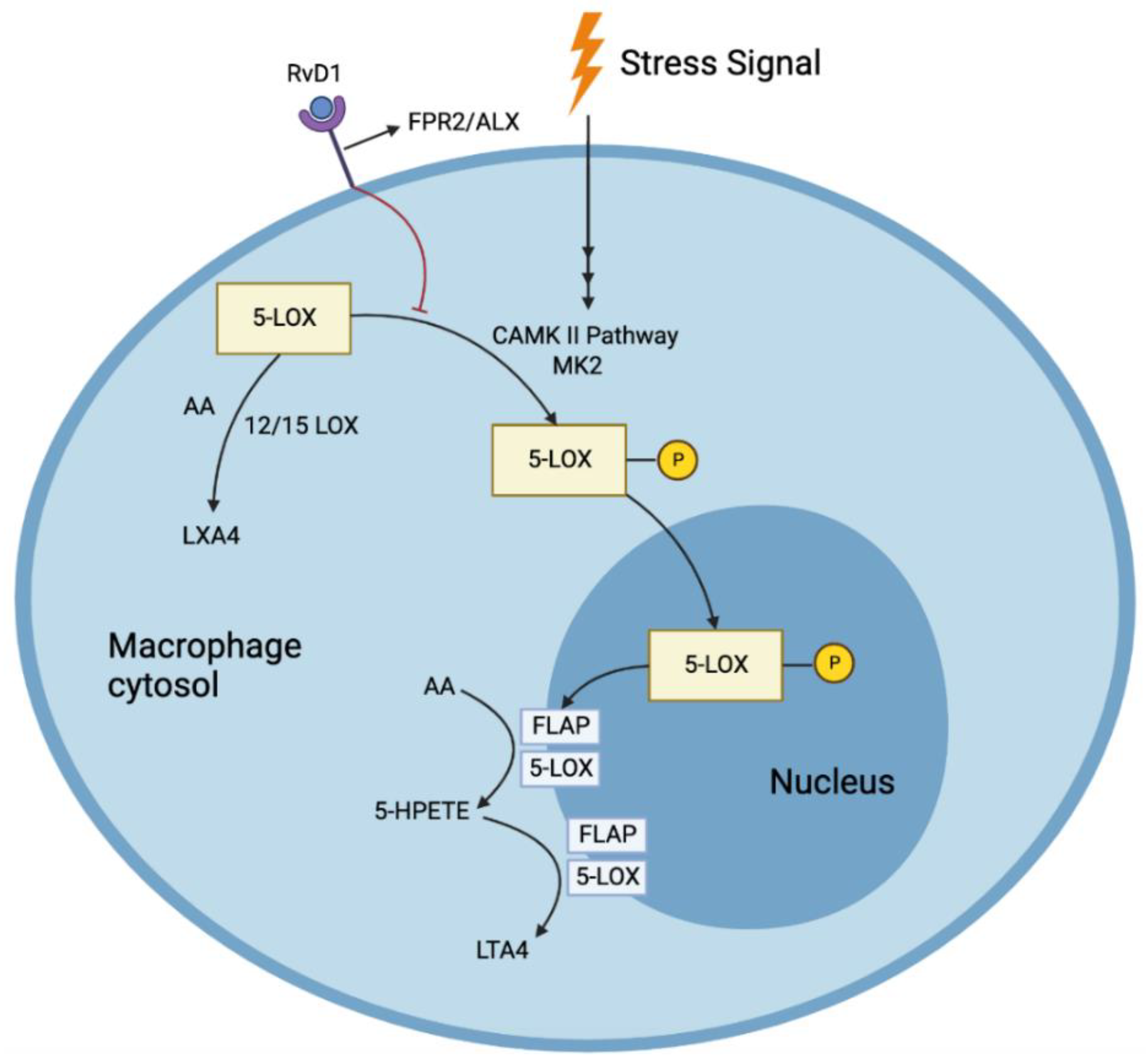
Figure 2. RvD1 prevents the nuclear location of 5-LOX, increasing the production of pro-resolving LXA4 and reducing the production of pro-inflammatory LTA4 in response to stress signals.
Liquid Chromatography tandem Mass Spectrometry of human carotid plaques revealed LXA4 and RvD1 as the major SPMs, requiring 5-LOX. Furthermore, these pro-resolving molecules were much less in vulnerable plaques than the stable ones. However, the intermediates such as 5-HEPE, 15-HEPE, 17, and 14-HDHA through the actions of 5/15/12-LOX were high in vulnerable plaques, indicating that the enzymes (5-LOX and 15-LOX) themselves were bioactive even in vulnerable plaques
[24]. One mechanism by which there could be a reduction in RvD1 was proposed to be the relocation of 5-LOX to the nucleus via the persistent activation of Ca
2+/Calmodulin Dependent Protein Kinase II (CAMKII) caused by the oxidative stress from substances such as 7-ketocholesterol (7-KC) found in the atherosclerotic plaque. This was proven by the reduction in the nuclear localization of 5-LOX once CAMKII expression was suppressed in human macrophages
[26]. Inhibition of CAMKII also prevented the reduction of RvD1 production by 7-KC
[24]. RvD1 also blocked the synthesis of LTB4 from AA. Such pathways were mediated by the receptors formyl peptide receptor 2/ lipoxin A4 receptor (FPR2/ALX), as both a receptor blocking antibody, as well as an antagonist, blocked this reduction of LTB4 by RvD1(
Figure 2) [26]. Nuclear localization of 5-LOX produces LTA4, which is then transformed to LTB4 through the action of LTA4 hydrolase. This nuclear localization is brought about by the phosphorylation of LOX-5 at Ser271 by p38MAPK-activated protein kinase 2 (MK2). RvD1 blocks this phosphorylation through the receptors FPR2/ALX and G-protein-coupled receptor (GPCR) Gi (and GPR32 in humans). LXA4, which shares the same receptors, also has been found to reduce the phosphorylation. RvD1 could not block the synthesis of LTB4 when incubated with large quantities of LTA4, demonstrating that RvD1 cannot alter the pathway once LTA4 is formed
[26]. Fredman et al. showed that the decrease in RvD1 was associated with the progression of the atherosclerotic plaque in the aortic arch of Ldlr−/− mice fed with a western diet for 8 or 17 weeks. Analysis of the lipid mediators in early vs. late plaques showed an approximately 87-fold decrease in RvD1 in advanced plaques but no significant change in LTB4
[24]. An increase in the lesional RvD1 levels was noticed when RvD1 was administered within the physiologic range. This increase resulted in the reduction of LTB4 levels, showing that RvD1 could have facilitated this reduction by preventing the nuclear localization of 5-LOX. This simultaneous increase in RvD1 (through external administration) and reduction in LTB4 (through RvD1 mediated reduction in the nuclear localization of 5-LOX) resulted in the ratio of RvD1:LTB4 being reverted to its early plaque levels. In addition, RvD1 increased other p SPMs, reduced oxidized CAMKII, reduced oxidative stress in the plaques, enhanced efferocytosis of macrophages, reduced the size of necrotic cores, and reduced the levels of collagenase and MMP9 without a concomitant reduction in the number of macrophages or VSMCs, resulting in a thickened fibrous cap. All these effects contribute to the stability of the plaque and in slowing its progression to an advanced type
[24]. RvD1 was shown to act on human PMN, in-vitro, through a GPCR receptor that was inhibited by Pertussis toxin (PTX) and reduced their actin polymerization. They also blocked β2 integrin molecules on human PMN that were regulated by LTB4. Enhancement of phagocytosis by macrophages was also observed through the interaction of the receptors ALX, GPR32, and RvD1
[27]. RvD1 was also found to limit monocyte adhesion, reactive oxygen species (ROS), and pro-inflammatory cytokine production in VSMCs derived from the saphenous vein in vitro and in rabbit arteries that underwent balloon angioplasty
[8]. It was found to alter the cytoskeletal properties of arterial smooth muscle cells (ASMC) in rats, thereby inhibiting their migration. Furthermore, it reduces their proliferation, oxidative stress, and translocation of p65, a molecule vital in NF-κB stimulation, which is implicated widely in the inflammation processes. All these beneficial effects were observed without damaging the viability of ASMCs
[5]. Interestingly, a positive effect of RvD1 on reducing neutrophil infiltration comes from the analysis done by Recchiuti et al. They found that RvD1, possibly through its upregulation of certain micro RNAs (miRNAs) in humans, brought about a reduction in the resolution interval by ~4 h. These miRNAs were found to target immune-competent proteins such as the NF-κB pathway and 5-LOX (in the leukotriene pathway). By blocking these pathways, the concentration of pro-inflammatory mediators is reduced
[28].
Akagi et al. demonstrated that pretreatment of ASMCsin vitro with DHA-derived SPMs, RvD2, and maresin-1(MaR1), impaired their migration towards PDGF in a dose-dependent manner by 74% and 80%, respectively at a 100 nM concentration
[29]. They also showed that GPCR could have mediated this response since the reduction in migration was attenuated in the presence of PTX, which inhibits GPCR proteins
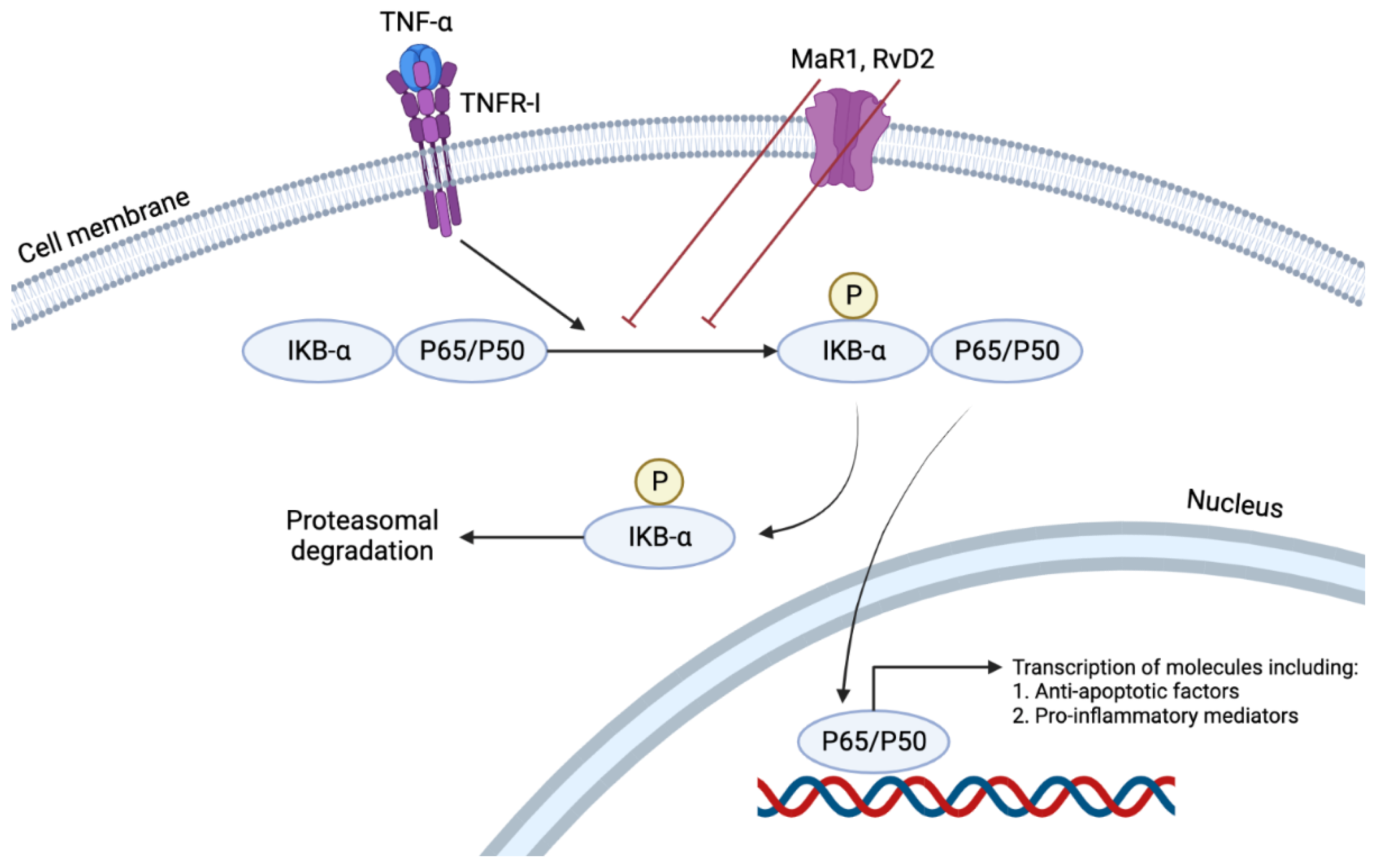
[8][27][29]. Of the many immunologically active cytokines, TNFα induces the NFκB pathway by nuclear translocation of p65 (
Figure 3). This pathway results in the transcription of many pro-inflammatory cytokines such as TNFα, Interleukin (IL)-1, IL-6, and IL-8. An in vitro study showed that RvD2 or MaR1 treatment of mouse ASMCs reduced p65 nuclear translocation by 24% and 28%, respectively, at a concentration of 500 nM. At the same concentration, such treatment also has reduced TNF-α induced superoxide production by 46% and 53%, respectively
[29]. Furthermore, RvD2 reduced the cultured VSMCs production of VCAM-1 and ICAM-1 induced by TNF-α
[8]. Such effects of these two SPMs demonstrate their anti-inflammatory and pro-resolving characteristics in their local environment. This pro-resolving action reduced the neo-intimal hyperplasia (neointima: media area ratio), which has been proven to be a result of chronic inflammation, by 67% and 71% by RvD2 and MaR1, respectively. The same study also showed a decrease in migration of neutrophils and monocytes to the area of injury, achieved by the suppression of MCP-1 expression by activated VSMCs and an increase in the M2 phenotype of macrophages which promoted resolution of the ongoing inflammation
[29].
Figure 3. TNFα induces the NFκB pathway by nuclear translocation of p65. This pathway results in the transcription of many pro-inflammatory cytokines such as TNFα, Interleukin (IL)-1, IL-6, and IL-8. RvD2 or MaR1 treatment reduces p65 nuclear translocation, demonstrating their anti-inflammatory and pro-resolving characteristics in their local environment.
Aspirin-triggered RvD1 (AT-RvD1) has been shown to activate the nuclear factor erythropoietin 2 related factor 2 (Nrf2), increasing the expression of genes such as heme oxygenase-1 (HMOX1), and NAD(P)H quinone oxidoreductase 1 (NQO-1), aiding in combating oxidative stress in mice lung injuries
[30].
7.2. Resovlin E Series (RvE)
Resolvin E (RvE) series (E1 and E2) molecules are produced from EPA through the action of 5-lipoxygenase (5-LOX)
[31]. In addition, RvE1 receptors have been found to be GPCRs
[32].
RvE series mediate the resolution of inflammation through the following means:
-
Reduction in chemotaxis of PMNs by affecting changes in their actin polymerization
[33].
-
Increase in non-phlogistic phagocytic activity of macrophages
[33].
-
Dose-dependent increase in IL-10 (an anti-inflammatory cytokine) by macrophages
[33].
-
Downregulation of leukocyte integrin activation, reducing their response to platelet activation factor (PAF), a potent pro-inflammatory cytokine. This downregulation was postulated to be mediated through the interaction of RvE2 and leukotriene B4 receptor, BLT1. RvE1 binds BLT1 and ChemR23 equally, but RvE2 is a weak agonist of the ChemR23 receptor
[33]. Platelet aggregation is one of the hallmarks of acute inflammation, brought about in part by ADP, which activates other platelets and leukocytes through intracellular signaling pathways. These pathways ultimately end with activation of platelet receptor GP IIb/IIIa, and granule secretion. RvE1 has been shown to regulate an ADP-mediated pathway that results in P-selectin surface mobilization through the ChemR23 receptor
[34].
These SPMs have proven to be strong local modulators of acute inflammation through these actions. One of the major risks in atherosclerosis—thrombus formation- is avoided by preventing platelet aggregation. GP IIb/IIIa is a receptor for fibrinogen that is activated by ADP within platelets. RvE1, at concentrations of ~100 nM, has reduced fibrinogen binding to platelets. The lack of complete blockage of ADP stimulation of platelets by RvE1 is beneficiary as platelet aggregation, and thrombus formation are required for hemostasis
[34].
 +1 credit
+1 credit