Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Ma, S.; Hou, Y.; Hao, J.; Lin, C.; Zhao, J.; Sui, X. Block Copolymers. Encyclopedia. Available online: https://encyclopedia.pub/entry/34104 (accessed on 21 July 2026).
Ma S, Hou Y, Hao J, Lin C, Zhao J, Sui X. Block Copolymers. Encyclopedia. Available at: https://encyclopedia.pub/entry/34104. Accessed July 21, 2026.
Ma, Shuhui, Yushuang Hou, Jinlin Hao, Cuncai Lin, Jiawei Zhao, Xin Sui. "Block Copolymers" Encyclopedia, https://encyclopedia.pub/entry/34104 (accessed July 21, 2026).
Ma, S., Hou, Y., Hao, J., Lin, C., Zhao, J., & Sui, X. (2022, November 11). Block Copolymers. In Encyclopedia. https://encyclopedia.pub/entry/34104
Ma, Shuhui, et al. "Block Copolymers." Encyclopedia. Web. 11 November, 2022.
Copy Citation
With the speedy progress in the research of nanomaterials, self-assembly technology has captured the high-profile interest of researchers because of its simplicity and ease of spontaneous formation of a stable ordered aggregation system. The self-assembly of block copolymers can be precisely regulated at the nanoscale to overcome the physical limits of conventional processing techniques. This bottom-up assembly strategy is simple, easy to control, and associated with high density and high order, which is of great significance for mass transportation through membrane materials.
block copolymer
self-assembly
nanochannels
1. Introduction
Two or more thermodynamically incompatible polymer blocks with diverse physical and chemical properties are covalently bonded together to form so-called block copolymers (BCPs) [1]. In the 1950s, the discovery of the surfactant Pluronic (PEO-b-PPO-b-PEO) [2][3][4] first attracted the attention of scientists. The mass production of BCP received a boost in 1956 due to the invention of living anionic polymerization [5]. Subsequently, other controlling polymerization techniques can also be used to synthesize BCP, such as living cationic polymerization [6], atom transfer radical polymerization (ATRP) [7][8][9][10][11], and reversible addition-fragmentation chain transfer polymerization (RAFT) [12][13]. Edwards’ self-consistent field theory (SCFT) [14] provided a premium theoretical tool for posterior simulations to explore the phase behavior of BCP. The foundations laid by these pioneers [15][16] have greatly facilitated the advancement of materials science, technology, and theory [17]. The chemical properties of various BCP blocks tend to vary with amenability to addition. To exploit the controllable properties of BCP, scientists have combined theoretical derivation with experimental practice to modify the structure of BCP for desired applications. This requires a thorough understanding of the factors that govern the nanostructure of BCP to regulate it according to the actual needs.
The BCP system can stand out and become the focus of research due to its incomparable self-assembly ability [18], and BCP can undergo accurate microphase separation in the range of 10–100 nm, which expands a new method to prepare nanodevices. The self-assembly of BCP forms rich nanostructures due to the difference in structure, block ratio, and sequences [19]. Surface structure and nanostructure are the keys to improving the properties in applications such as electronic lithography [20], organic photovoltaics [21], nanofiltration [22], and ultrafiltration [23], where electron transport or surface features dictate the performance of the devices. BCP has the potential to boost the properties of these nanodevices [24], which can result in their flourishing applications in different areas due to unique nanoscale customizable template nanostructures [25]. Researchers intend to highlight the effective exploitation and performance enhancement of BCP for novel energy sources instead of repeating the aforementioned applications. Researchers' focus will be on the construction and application of BCP-based nanostructured materials in mass transport for energy conversion. The massive exploitation and usage of nonrenewable resources, such as oil and coal, has become a burden on the environment [26]. In recent years, global warming has been responsible for frequent natural disasters [27][28], which have resulted in an irresistible surge to limit the traditional energy methods that incur huge carbon emissions and foster innovative and efficient clean energy sources. Concentration cells, fuel cells, and rechargeable batteries that can convert chemical energy into electricity without carbon emissions are new energy sources with the potential to replace traditional energy sources in the foreseeable future [29]. Using membranes for mass transportation is a key part to obtain high power efficiency from the cells of these devices related to new energy conversion processes [30], which makes the development of a high-performance battery diaphragm very important.
2. Microphase Separation of Block Copolymer
Spontaneous phase separation occurs in BCP due to repulsion between chemically and thermodynamically incompatible blocks. At the macroscopic scale, the phase separation between blocks cannot occur due to the presence of covalent bonds between the blocks of BCP [31]. Instead, in the macromolecular-length scale, phase separation can only occur at the microscopic scale, i.e., microphase separation [32]. Due to the microphase separation, there is structural arrangement of BCP at the macroscopic scale, which is known as the self-assembly phenomenon [33] and responsible for the generation of new boundaries between blocks and can result in various assembly structures.
Three significant parameters are responsible for the microphase separation between the blocks: (1) the relative volume fraction f of each block, which is used to characterize the microscopic composition of BCP; (2) the Flory–Huggins parameter χ, which is used to characterize the interaction between the blocks [34][35] and is inversely proportional to the temperature change; (3) the total degree of polymerization N of the polymer. Among them, χN, which represents the phase separation strength, plays a decisive function in the separation state of BCP [36]. Increasing the temperature or decreasing χN gradually decreased the incompatibility between the blocks. Molecular-level mixing changes polymeric molecular chain from the stretched chain state with obvious interfaces and ordered arrangement to the disordered Gaussian chain state, as shown in Figure 1a, which results in an order-to-disorder transition (ODT) [37] of the polymer. The critical χN value for the occurrence of ODT is approximately 10.5. Electron microscopy can be used to clearly observe the ODT transition process of some BCPs with lower molecular weights, as shown in Figure 1b, which shows TEM images of poly(styrene)-b-poly(imide) (PS-b-PI) before and after undergoing the ODT process.
Due to the introduction of new theories such as the thermal up-and-down effect and mean-field theory, the improved self-consistent field theory (SCFT) can more accurately calculate the BCP phase behavior by scientists [38]. The theoretical phase diagram of the AB diblock copolymer [39] based on self-consistent mean-field (SCMF) theory showing the changes in phase behavior with increasing or decreasing values of fA and χN in a concrete manner is shown in Figure 1c (left). However, the experimental phase diagram deviates from the theoretical phase diagram because there are various practical and objective factors. Figure 1c (right) shows the experimental phase diagram of the PI-b-PS copolymer obtained by Bates et al. Both experimental and theoretical phase diagrams of PI-b-PS are qualitatively consistent with the variation occurring on the quantitative scale. First, the critical χN value for ODT is approximately 20, with the loss of symmetry for the phase diagram at approximately fA = 1/2. Instead of the CPS phase, a mesostable porous lamellar PL phase appears between C and L regions (calculations confirmed the mesostability of this phase later on). The difference in the PI and PS blocks from the theoretically assumed AB blocks is the main reason behind these deviations. The molecular morphology, properties, and interactions between PI and PS are responsible for the difference in experimental and theoretical values. In the experimental phase diagram, the ODT transitions can directly proceed in both the disordered D region and ordered regions that only occur at the critical point in the theoretical phase diagram, and this phenomenon can be mainly attributed to the effect of the thermal up-and-down phenomenon. With an increasing number of blocks, the phase behavior of block copolymers becomes more complicated [40][41][42].
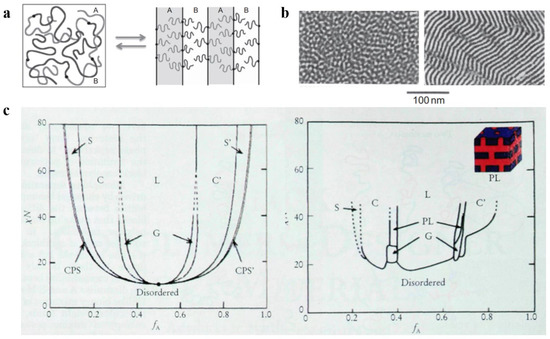
Figure 1. (a) Molecular chain distribution of AB-type diblock copolymer before and after the ODT [37]. (b) TEM images of PS-b-PI before and after the ODT [37]. (c) Theoretical phase diagrams predicted by the self-consistent field theory of the linear AB diblock copolymer and the actual phase diagram (spherical (S, S’), cylindrical (C, C’), bicontinuous gyroidal (G), and lamellar (L)) obtained from the PS-b-PI [39].
3. Regulation of the Self-Assembled Structure of the Block Copolymer
3.1. Influence of the Molecular Structure on the Self-Assembled Structure
The structural morphology of BCP films is the main interest. The self-assembly of BCP film is affected by many factors [43][44][45] which can be divided into molecular structure factors and assembly condition factors. The most fundamental strategy to regulate the nanoscale morphology of BCP is to adjust its molecular structure, since the BCP structure determines the self-assembly properties. Above the ODT transition curve, the thermodynamically stable nanostructures of BCP mainly consist of spherical S, cylindrical C, bicontinuous gyroidal G, and lamellar L geometries. As shown in Figure 2a, a change in volume fraction f of the AB block changes the morphology of the AB diblock copolymer [46]. Depending on whether fA < 0.5 or fB < 0.5, block A is dispersed within the continuous phase composed of block B or vice versa, and with increasing fA, the transformation of S→C→G→L occurs, while the same S’→C’→G’→L transition occurs with increasing fB. The size of each block in each form is determined by the molecular weight and compatibility of the blocks. With increasing molecular weight of the polymer, the blocks become segregated with increasing distance between the domains, and these factors make it challenging to undertake studies of high molecular weight BCPs [47].
The structure of BCP is also characterized by an essential parameter known as polydispersity index (PDI) [48] of molecular weight, which is calculated as PDI = Mn/Mw [49]. The BCPs obtained from the polymerization of reactive anions are monodisperse [50]. However, in recent years, with the emergence of novel methods for synthesizing BCPs, most of the obtained BCPs have been polydisperse [51]. It is necessary to study the effect of broadening the molecular-weight distribution on the phase behavior of BCPs. With the concept of maintaining thermodynamic equilibrium to the maximum possible intent, Ruzette et al. [52] investigated the self-assembly behavior of triblock copolymer ABA with varying molecular-weight distributions, with narrower dispersion of block B (poly (butyl acrylate) (PBA)) and wider dispersion of block A (poly (methyl methacrylate) (PMMA)). The resulting transparent BCP membranes are homogeneous. The TEM characterization reveals the possibility of forming classical S, C, G, and L structures. However, a highly curved interface between the blocks and the PMMA block was formed mainly due to the shift of the interface to release the stretching energy of the molecular chains.
Lynd et al. [53] investigated the effect of molecular-weight distribution on the occurrence of ODT for two different BCPs prepared of polydispersed poly (ethylene-acrylamide)-b-poly (DL-propyleneglycol) (PL) and polystyrene-b-polyisoprene (SI). Ultimately, during the occurrence of ODT, increasing polydispersity in blocks with fewer (f < 0.5) or more (f > 0.5) components decrease or increase in the critical χN value, respectively. Widin et al. [54] reported the structural effects of a low-dispersion block polystyrene (PS) and a high-dispersion block polybutylene (PB) on the triblock copolymer SBS. As shown in Figure 2b (left), the molecular chain of highly dispersive block PB is stabilized by the low dispersive PS block in the middle position. Finally, similar experimental results to those of Ruzette were obtained, where the phase separation interface was bent toward the polydisperse segment because the polydisperse block has a smaller filling volume than the monodisperse block of the same molecular weight (the phase interface bending process is shown in Figure 2b (right)). The polydisperse B-block also reduces the critical χN value for ODT and increases the position of the lamellar phase window. The investigations show that by playing with the polydispersity of a particular block in BCP, the microphase separation state of BCP and its thermodynamic stability can be regulated.
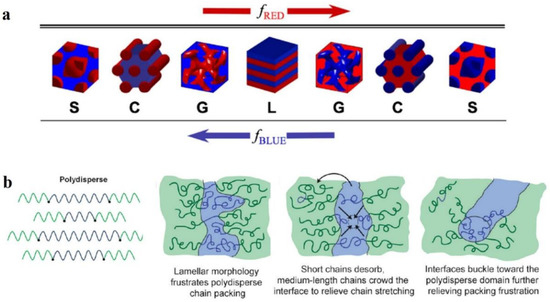
Figure 2. (a) Four equilibrium-ordered state transitions in diblock copolymers determined by relative volume fraction f. Spherical (S), cylindrical (C), bicontinuous gyroidal (G), and lamellar (L) [46]. (b) Schematic diagram of polydispersed B-blocks in ABA-type triblock copolymers leading to distortion of block microdomain interfaces [54].
3.2. Influence of Assembly Conditions on the Self-Assembled Structure
Normally, in addition to the interactions between polymer molecules, external film formation conditions can also affect the nanostructure. Hence, the type of solvent, polymer solution concentration, nonsolvent, additives, and external field conditions during the film formation process can affect the structure of BCP.
3.2.1. Solvents to Prepare the Membranes and Self-Assembly
The selectivity of solvents toward different blocks significantly affect the self-assembled structures. Hanley et al. [55] dissolved PS-b-PI in bis(2-ethylhexyl) phthalate (DOP), which is a nonselective solvent for PS and PI, di-n-butyl phthalate (DBP), and diethyl phthalate (DEP) solvents, which are sequentially more selective for the PS block, and tetradecane (C14), which is a selective solvent for the PI blocks, to observe their phase behavior by small-angle X-ray scattering (SAXS) (Figure 3a). The PS-b-PI membrane exhibited the transformation of G→hexagonal filled C→D, which, when dissolved in different solvents, exhibited different nanostructures. After dissolution in DBP, the phase transition process of PS-b-PI was L→G’→C’→S’. The phase behavior in DEP, which is more selective for PS, like DBP, was similar to that of DBP but with higher TODT. In contrast, in C14, for the PI selective solvent, the G’→C’→S’ ordered phases were the only occurring transitions because enhancement of segregation between microdomains by the selective solvents improve the stability of the ordered phase, which results in the appearance of more ordered phase behavior. The formation and closure of pores in BCP nanostructures can also be regulated by selective solvents [56]. The spontaneously formed poly(styrene)-b-poly (2-vinyl pyridine) (PS-b-P2VP) membrane is a dense, nonporous film. However, when dissolved in P2VP selective solvent (e.g., ethanol), the P2VP blocks dissolve to form a membrane with round or elliptical pores surrounding the core of the PS blocks. With increasing time or temperature, these pores grow into a columnar network and finally form three-dimensional interconnected pores. This pore formation process is often reversible, and immersion of the porous membrane in a PS block selective solvent (e.g., cyclohexane) closes the pores to restore the dense, nonporous membrane.
In addition, dissolution of the self-assembled structure of BCPs in mixed solvents should be explored. Yi et al. [57] investigated the nanostructure of poly(styrene)-b-poly (4-vinyl pyridine) (PS-b-P4VP) by adding it to N, N-dimethylformamide, DMF, (selective for P4VP block, and slow volatilization), tetrahydrofuran, THF, (selective for PS, faster volatilization), and their mixtures. In pure DMF solvent, a nanoscale spherical structure was obtained for PS-b-P4VP, a dense and smooth surface was obtained in pure THF, while the mixed solvent yielded a rod-like aggregated structure where the diameter of the nanospheres was similar to that observed in DMF. When polymers are dissolved in a mixture of solvents with different solubilities and volatilities, the preferential evaporation of one solvent leads to a concentration gradient within the BCP. This concentration gradient leads to phase separation of the BCP, the so-called evaporation-induced phase separation (EIPS) [58] of the solvent.
In addition, the concentration of the polymer solution is an important factor that affects the nanostructure of BCP. Jiang et al. [59] was the first to simulate the membrane formation of a double hydrophobic poly(styrene)-b-poly (methyl methacrylate) (PS-b-PMMA). The effect of the polymer concentration on the morphology of polymer molecules was also investigated in the polymer concentration range of 10–55%. Figure 3b clearly shows the difference in morphologies corresponding to different polymer concentrations. For low polymer concentrations, microphase separation just begins to occur, and the formation of a continuous phase in the system under this condition is not possible because there is not a sufficient BCP concentration, which forms a spherical structure at lower concentrations. With increasing polymer concentration, a structure can be observed because a continuous phase forms by BCP. With a further increase in concentration, porous membranes with elliptical pores start to form. When the concentration increases to approximately 30%, the elliptical pores change to circular pores. From there on, a further increase in polymer concentration only decreases the pore size within the porous membrane and membrane wall thickening. When the polymer concentration reaches approximately 45%, the polymer shows an irregular sponge-like, cross-linked structure, and finally, a continued increase in polymer concentration causes the pores to plug and results in a continuous structure of the polymer. This experiment indicates that, within a certain concentration range, the polymer concentration can be adjusted to achieve different polymer morphologies and obtain porous membranes with ideal pore size and stability.
Formation of the BCP membrane by selective solvents requires three steps: solvent uptake, swelling, and solvent evaporation drying. The effects of the first two steps on the BCP have been described in the previous section, while the evaporation rate of the solvent has been found to affect the structural orientation of BCP. It has been proven that fast solvent evaporation leads to a vertical orientation of the internal structure [60], while slower solvent evaporation rates cause parallel structural orientation or structures with mixed orientation [61]. The evaporation rate determines the propagation of phase separation from the membrane surface to the interior. Similar conclusions were obtained by Phillip et al. [62] when comparing the morphology of poly(styrene)-b-poly (D, L-lactide) (PS-b-PLA) under rapid solvent evaporation and slow evaporation (Figure 3c). Because ultrafast solvent evaporation makes steep concentration gradients rapidly propagate the vertical direction of the membrane surface, the cylindrical structure orients along the surface’s normal direction.
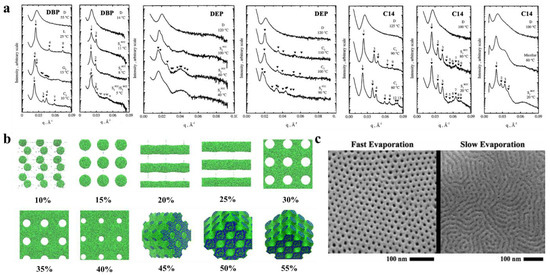
Figure 3. (a) Representative SAXS profiles as a function of temperature for DBP, DEP, and C14 solutions [55]. (b) Nanoscale morphology diagram of PS-b-PMMA porous membrane simulated by DPD when dissolved in tetrahydrofuran solvent at different polymer concentrations. PMMA is shown in green, and PS is shown in blue [59]. (c) SEM images of different morphologies formed by PS-b-PLA at varied solvent evaporation rates [62].
During the self-assembly process via solvent vapor annealing (SVA), the solvents are found to have a significant influence. For a given polymer system, the choice of different solvents can result in different nanostructures [63][64][65]. Peng et al. [66] investigated the effect of SVA on the morphology of poly(styrene)-b- poly (ethylene oxide) (PS-b-PEO). As shown in Figure 4a, the nanostructure of the PS-b-PEO membrane obtained by conventional casting at room temperature was a disordered and irregular worm-like morphology, while the PS-b-PEO membrane resulting from 5 min of steam annealing treatment with toluene (a selective solvent for PS) was transformed into a highly ordered arrangement of hexagonally filled perpendicular cylindrical microdomains.
However, due to preferential wetting of the surface, a thermodynamically preferred morphology should be formed with a cylindrical orientation parallel to the surface instead of perpendicular to the surface, as experimentally obtained in [67]. The formation mechanism of this morphology has not been understood, and various researchers have attempted to use computer simulations or experimental investigations to explain this phenomenon [68][69][70]. Phillip et al. [71] analyzed the evolution of the internal morphology of PS-b-PLA during SVA. Theoretical analysis and experimental verification found that the generation of such cylinders evolved from spherical intermediates nucleated at the vapor/polymer membrane interface due to the rapid evaporation of solvent, which created a concentration gradient in the BCP, and the perpendicular orientation of the intermediates with respect to the surface and epitaxial growth into cylinders. However, too-fast evaporation of the solvent does not allow sufficient time for nucleation. Lin et al. [72] used TEM analysis to demonstrate that the length of this ordered arrangement could reach the level of membrane thickness. In other words, SVA can achieve a directional ordered arrangement perpendicular to the membrane surface throughout the membrane, which is of great significance for the development of certain ion- or proton-conducting membranes or water-permeation membranes with controlled pore sizes. Kim et al. [73] demonstrated that SVA yielded a more ordered, structurally stable membrane than those obtained by conventional methods by imparting the molecular chain mobility, shielding the surface energy of the two blocks at the vapor/BCP interface and the interfacial energy between polymer and substrate, and rapidly eliminating defects in the BCP structure (Figure 4b).
When the blocks in BCP strongly interact, more variables in the SVA process will affect the structure [65][74][75]. For instance, Park et al. [76] proposed structural changes in a PS-b-PMMA membrane by choosing a selective or neutral substrate (different substrate treatments with the generated SEM images of varying morphologies are shown in Figure 4c). The membranes on neutral substrates exhibit a cylindrical structure perpendicular to the surface at the top (near the air) and bottom (near the substrate). For the SVA process on selective substrates, the top structure of the BCP membrane was unaffected, exhibiting a cylindrical structure perpendicular to the surface, but the bottom structure changed to a cylindrical structure parallel to the substrate. This result demonstrates the possibility of tailoring the pattern and structure of the BCP membrane by adjusting the membrane–substrate interaction. Cheng et al. [77] investigated various process parameters during the SVA process and ultimately demonstrated that the temperature of the substrate and vapor were also key factors in controlling the structural morphology of BCP membranes.
However, the ordered BCP membrane obtained by the selective solvent SVA method is only short-ranged. It has been shown that long-range ordering of BCP membrane in the lateral direction can be obtained via annealing with nonselective solvent (solvent with the same selectivity for each block and no specific interactions) vapors during SVA [78]. Bosworth et al. [79] obtained a long-range ordered BCP membrane by annealing poly(α-methylstyrene)-block-poly(4-hydroxystyrene) in acetone (selective solvent) vapor, which resulted in a spherical structure and tetrahydrofuran (nonselective solvent) vapor, forming a columnar phase parallel to the surface. This is due to the fact that the nonselective solvent is only uniformly distributed between the molecular chains and same selectivity for each block. It increases only their mobility without any other interactions, resulting in spontaneous formation of a cylindrical orientation parallel to the membrane plane with the lowest surface energy. SVA overcomes the shortcomings of conventional thermal annealing and enables highly ordered rearrangement of BCP in a relatively short time. By reducing the effective Tg, it weakens the interactions between chains and between chains and substrates to increase the mobility of polymer chain [80].
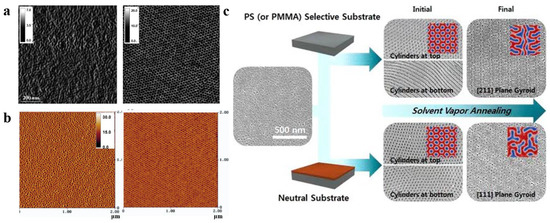
Figure 4. (a) AFM phase images (1 × 1 μm2) of as-cast conventional PS-b-PEO film and PS-b-PEO film annealed in PS-selective toluene vapor for 5 min [66]. (b) AFM phase diagrams of PS-b-PEO films obtained by spin-coating method and annealed in neutral benzene vapor for 48 h [73]. (c) SEM images of the (111) and (211) planes produced by solvent vapor annealing of PS-b-PMMA films in neutral substrate and selective substrate [76].
3.2.2. Nonsolvent Approaches for Constructing Nanostructure
Researchers have also investigated the effect of nonsolvent-induced phase separation (NIPS) [81] of BCPs intensively [82][83][84]. The polymer is dissolved in solvent to form a homogeneous solution, followed by the slow addition of reagents which are more soluble in the solvent compared to the polymer (called extractants) to extract the solvent. The resulting two-phase structure with the polymer as continuous phase and solvent as the dispersed phase can lead to a polymer with a certain pore structure after the removal of solvent. Plisko et al. [85] modified poly (ethylene glycol)-b-poly (propylene glycol)-b-poly (ethylene glycol) (Pluronic F127) by the NIPS method employing polysulfone (PSF), and the polymer membrane obtained by the NIPS method was found to be porous in comparison to the dense polymer membrane prepared by the EIPS method (the surface structure of the porous film is shown in Figure 5a). Similarly, Gu et al. [86] fabricated porous PI-b-PS-b-P4VP membrane with sponge-like structure using the NIPS method, and Figure 5b shows the cross-sectional structure of the sponge-like porous film. NIPS is the most commonly used method for the preparation of membranes for separation [87][88], but the pore size and distribution of pores on the surface of NIPS-formed membranes are irregular and not well-controlled [89][90], and presence of some large pores can lead to material defects resulting in reduced mechanical strength of the membrane. This feature is responsible for limiting the application of BCP membranes made by the NIPS method in a great way.
In order to overcome the shortcoming mentioned above, a new NIPS strategy has emerged in the recent years: a short self-assembly is formed via short solvent evaporation after pouring the BCP into a solvent or mixed solvent, and then it was immersed in a nonsolvent for NIPS [91][92]. By this method, a monolithic, asymmetrically structured membrane, as shown in Figure 5c,d, can be formed [93]. Self-assembly results in the formation of highly ordered, dense pores in the upper thin selective layer which are uniformly perpendicular to the membrane surface [94]; pores of the lower layer which exhibit a sparse, disordered sponge-like interconnected pore channel are formed by NIPS [95]. This method of combining self-assembly (S) of BCP with nonsolvent-induced phase separation (NIPS) is named SNIPS [96][97]. As the first step of membrane formation via SNIPS is dissolution of BCP for self-assembly, different solvent parameters that affect the nanostructure (different selective solvents, mixed solvents, and solvent volatilization rate) discussed in the previous section also affect the membrane structure formed by the SNIPS method [98][99][100].

Figure 5. (a) SEM image of the surface of porous ultrafiltration membrane formed by the NIPS method for Pluronic F127 [85]. (b) SEM image of cross-section of multiporous sponge-like structure PI-b-PS-b-P4VP formed by NIPS method [86]. SEM images of the surface (c) and cross section (d) of PS-b-P4VP membrane obtained by SNIPS [93].
3.2.3. The Influence of External Field on Self-Assembled Structure of BCP
Thermal annealing has proved to be one of the efficient methods for preparing long-range ordered BCP membranes [101][102][103][104], and temperature is an effective and adjustable experimental condition during the process of self-assembly. Thermal annealing refers to the heating of BCP above its glass transition temperature Tg [105] for allowing the molecular chains within the polymer to self-assemble to the state closest to thermodynamic equilibrium [106][107][108]. Stehlin et al. [109] used AFM to observe the morphological variation of PS-b-PMMA at 180 °C and 200 °C (well above its Tg). Their resultant AFM images are shown in Figure 6a. It illustrates that thermal annealing results in ordered arrangement of the molecular chains, and the orderliness of PS-b-PMMA structure at 200 °C is higher than that of 180 °C for the same time, which indicates that the higher the annealing temperature, the faster the order of the arrangement. However, the BCP membrane produced by conventional thermal annealing techniques often shows structural defects, and the range of order regulation is limited, and for a thick membrane, ordering of the molecular chains over a large area cannot be obtained [110]. Moreover, for some polymers, the spacing between Tg and the viscous flow temperature Tf is small, which may not be sufficient for the rearrangement of molecular chains to take place within a short time.
In some systems, a combined method of the thermal annealing and solvent annealing methods can be used [111]. Kim et al. [112] experimentally investigated PS-b-P4VP membrane in chloroform vapor (the schematic diagram is shown in Figure 6b). Since chloroform is selective for the PS block, the PS block is pulled toward the outside of the membrane, and the P4VP block forms multilayer spherical micelles. The solvent vapor increases the mobility of the polymer molecular chains by swelling the membrane, and consequently, some of the P4VP blocks fuse. A single SVA process is insufficient to drive the fusion of P4VP spherical structures and subsequently convert them into columnar structures perpendicular to the membrane surface as a result of strong repulsion between the PS and P4VP blocks (B). During the cooling process, chloroform vapor escapes from the inside along the direction perpendicular to the membrane as the membrane shrinks, which allows the spherical structures to aggregate along the vertical direction (C). During further SVA with programmed heating, the aggregation of spherical microdomains can propagate to the inner center of the film (E), resulting in membrane expansion (D), and shrink when cooled. After several cycles of operation, vertically oriented P4VP cylindrical microdomains are obtained (F). This experiment provides an effective orientation method for the orderly arrangement, with strong interactions between the blocks or thick membrane. Furthermore, combining thermal annealing with SVA is enabled to surmount the structural uncontrollability difficulties of high molecular weight BCP, which is due to the low mobility of molecular chains caused by chain entanglement [113].
As early as 1991, Gurovich [114] made a theoretical derivation of the force and phase behavior of BCP in an electric field, but application of electric fields in the research of BCP has stopped at the theoretical level for a while since then. This delay ended when A. Bo¨ker [115] observed the morphological changes of due to the application of DC electric field on the PS-b-PI prepared via reactive anion polymerization. In the absence of an applied electric field, the PS-b-PI self-assembled as ordered lamellar structures in the control group, while under the application of an electric field, lamellar structures of the samples underwent macroscopic orientation along the electric field lines. The application of electric field achieves long-range ordered nanostructures. Schmidt [116] verified the changes in the lamellar structure of PS-b-PI with increasing strength of the electric field. The phase space parallel to the electric field direction decreases, while the phase space perpendicular to the electric field direction increases, mainly resulting from the stretching of polymer molecular chains along the electric field lines (the mechanism is shown above in the Figure 6c). Schoberth et al. [117] used quasi in situ scanning force microscopy (SFM) capable of solvent vapor treatment of BCP despite high electric fields to characterize the phase behavior. Figure 6c below shows the SFM image of the disordered Polystyrene-b-poly (2-hydroxyethyl methacrylate)-b-poly (methyl methacrylate) S47H10M43 oriented along the electric field direction after the application of an electric field [118]. Subsequently, it was experimentally demonstrated that application of an electric field decreases the TODT of PS-b-PI [119].
BCP orientation can also be regulated by the application of shear fields [120]. Unidirectional shear force is applied on the BCP membrane by contacting with suitably selected moving solid surface to induce the internal structure transformation of BCP. The shear modification of polystyrene-b-polybutadiene-b-polystyrene (SBS) and the study of its rheological behavior were carried out by Morrison et al. [121]. The structure of SBS can become oriented along the direction of shear force. Angelescu et al. [122] demonstrated that the rate of shearing plays a decisive role for obtaining the molecular alignment. Low shearing rates allow the molecular chains to arrange in an ordered manner along the shear direction, while too-high shear rates may cause a disordered arrangement of the intramolecular structures. It was then shown that for similar temperature, shear orientation requires less time compared to annealing and generates structures with other defects such as disruption of grain boundaries and absence of dislocations almost in the shear range [123]. Pujari et al. [124] observed a lamellar structure perpendicular to the substrate when the self-assembly of BCP was obtained through the application of shear stress to PS-b-PMMA during annealing. Different structures of BCP such as S, C, and L structures were oriented through the application of shearing. Davis et al. [125] investigated the morphological changes of poly(styrene)-b-poly (n-hexyl methacrylate) (PS-b-PHMA) due to application of shearing and also probed the effect of shearing on different polymer composition (see Figure 6d for a schematic diagram of the apparatus for applying shear and its results). It was finally concluded that shear causes orientation along the shear direction, and that for membranes with varying PS content, the dislocation density is maximum for the membrane containing the highest amount of PS. The ordering of shear field presents a fresh approach toward nanostructural adjustment of BCP membrane, which can achieve unprecedented structural ordering at lower temperatures and under simpler conditions [126][127].
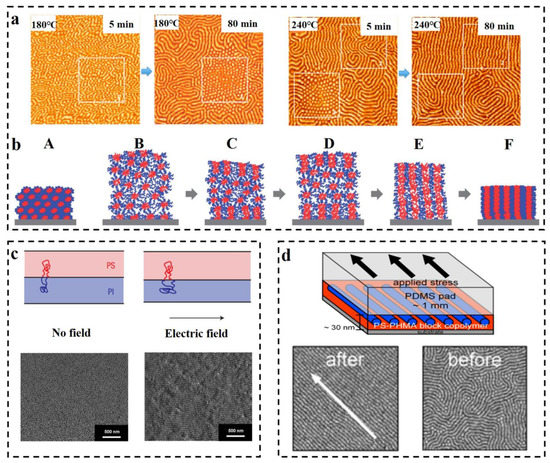
Figure 6. (a) In situ AFM images (1.25 × 1.25 μm2) of PS-b-PMMA obtained by thermal annealing at 180 °C and 240 °C for 5 min and 80 min, respectively [109]. (b) The cylindrical microdomain structure of PS-b-P4VP perpendicular to the surface is illustrated by the combination of programmed temperature rise and fall and SVA [112]. (c) The impact of electric field on the stretching of BCP molecular chains is illustrated and the microdomains are macroscopically oriented along the electric field direction in SEM images [116][118]. (d) Schematic diagram of the unidirectional shear force applied to PS-b-PHMA and TM-AFM phase images of the block nanoscale before and after shear force application. Scale bar: 500 nm [125].
3.2.4. Additives for Regulating Self-Assembly Structure
Researchers also explored various types of substances that can act as electrostatic additives [128] to regulate the nanomorphology of BCP [129], especially, to obtain some morphology in narrow interval [130]. Ionic liquid (IL) is a liquid composed of only anions and cations, with high ionic conductivity, chemical stability, and high thermal stability. Ionic liquids, when doped into BCP, can selectively complex with the charged blocks and induce lyotropic phase transitions via strong electrostatic cohesion [131]. Kim et al. [132] impregnated BCP into different ionic liquids to observe the variation in morphologies. It was demonstrated that the IL, 1-ethyl-3-methylimidazolium p-toluenesulfonate, could induce the structural transformation of the BCP from L to C morphology at a particular concentration, and different ILs result in the induction of different morphologies. Later, Bennett [133] added the IL 1-ethyl-3-methylimidazolium bis(trifluoromethanesulfonyl)imide (EMIM) to PS-b-PMMA to obtain a phase transition in the following sequence: D→L→G→C→D→S. IL-induced lyotropic phase transitions of BCP correspond to the single ion-induced phase transitions, while the role of multicharged ions on the phase transitions have rarely been reported, although it is easier to obtain strong electrostatic cohesion by multicharged ions. Polyoxometalate (POM) is a class of polymetallic oxygen cluster compounds formed by pretransition metal ions linked by oxygen, which has recently been applied for controlling the BCP nanostructures [134][135]. Zhang et al. [136] observed a change of an originally ordered columnar phase structure of the block copolymer into a disordered bicontinuous phase structure via incorporating Keggin-type POM H4SiW12O40 (SiW) into PS-b-P4VP, which was due to strong electrostatic cohesion between the POM and P4VP chains.
References
- Schacher, F.H.; Rupar, P.A.; Manners, I. Functional Block Copolymers: Nanostructured Materials with Emerging Applications. Angew. Chem. Int. Ed. 2012, 51, 7898–7921.
- Bates, C.M.; Bates, F.S. 50th Anniversary Perspective: Block Polymers-Pure Potential. Macromolecules 2017, 50, 3–22.
- Mankowich, A.M. Micellar Molecular Weights of Selected Surface Active Agents. J. Phys. Chem. 1954, 58, 1027–1030.
- Vaughn, T.H.; Suter, H.R.; Lundsted, L.G.; Kramer, M.G. Properties of some newly developed nonionic detergents. J. Am. Oil. Chem. Soc. 1951, 28, 294–299.
- Szwarc, M.; Levy, M.; Milkovich, R. Polymerization initiated by electron transfer to monomer. A new method of formation of block polymers. J. Am. Chem. Soc. 1956, 78, 2656–2657.
- Lanson, D.; Ariura, F.; Schappacher, M.; Borsali, R.; Deffieux, A. Application of living ionic polymerizations to the design of AB-type comb-like copolymers of various topologies and organizations. Macromol. Res. 2007, 15, 173–177.
- Charleux, B.; Delaittre, G.; Rieger, J.; D’Agosto, F. Polymerization-Induced Self-Assembly: From Soluble Macromolecules to Block Copolymer Nano-Objects in One Step. Macromolecules 2012, 45, 6753–6765.
- Nasrullah, M.J.; Vora, A.; Webster, D.C. Block Copolymer Synthesis via a Combination of ATRP and RAFT Using Click Chemistry. Macromol. Chem. Phys. 2011, 212, 539–549.
- Siegwart, D.J.; Oh, J.K.; Matyjaszewski, K. ATRP in the design of functional materials for biomedical applications. Prog. Polym. Sci. 2012, 37, 18–37.
- Matyjaszewski, K. Advanced Materials by Atom Transfer Radical Polymerization. Adv. Mater. 2018, 30, 1706441.
- Keddie, D.J. A guide to the synthesis of block copolymers using reversible-addition fragmentation chain transfer (RAFT) polymerization. Chem. Soc. Rev. 2014, 43, 496–505.
- Eggers, S.; Eckert, T.; Abetz, V. Double thermoresponsive block-random copolymers with adjustable phase transition temperatures: From block-like to gradient-like behavior. J. Polym. Sci. Pol. Chem. 2018, 56, 399–411.
- Gyorgy, C.; Hunter, S.J.; Girou, C.; Derry, M.J.; Armes, S.P. Synthesis of poly(stearyl methacrylate)-poly(2-hydroxypropyl methacrylate) diblock copolymer nanoparticles via RAFT dispersion polymerization of 2-hydroxypropyl methacrylate in mineral oil. Polym. Chem. 2020, 11, 4579–4590.
- Edwards, S.F. The statistical mechanics of polymers with excluded volume. Proc. Phys. Soc. 1965, 85, 613–624.
- Matsen, M.W.; Schick, M. Stable and unstable phases of a diblock copolymer melt. Phys. Rev. Lett. 1994, 72, 2660–2663.
- Matsen, M.W.; Bates, F.S. Unifying weak- and strong-segregation block copolymer theories. Macromolecules 1996, 29, 1091–1098.
- Spencer, R.K.W.; Matsen, M.W. Fluctuation effects in blends of A plus B homopolymers with AB diblock copolymer. J. Chem. Phys. 2018, 148, 204907.
- Kang, S.; Kim, G.-H.; Park, S.-J. Conjugated Block Copolymers for Functional Nanostructures. Acc. Chem. Res. 2022, 55, 2224–2234.
- Rosler, A.; Vandermeulen, G.W.M.; Klok, H.A. Advanced drug delivery devices via self-assembly of amphiphilic block copolymers. Adv. Drug Deliv. Rev. 2012, 64, 270–279.
- Cummins, C.; Lundy, R.; Walsh, J.J.; Ponsinet, V.; Fleury, G.; Morris, M.A. Enabling future nanomanufacturing through block copolymer self-assembly: A review. Nano Today 2020, 35, 100936.
- Meng, L.; Fan, H.; Lane, J.M.D.; Qin, Y. Bottom-Up Approaches for Precisely Nanostructuring Hybrid Organic/Inorganic Multi-Component Composites for Organic Photovoltaics. MRS Adv. 2020, 5, 2055–2065.
- Yu, H.Z.; Qiu, X.Y.; Moreno, N.; Ma, Z.W.; Calo, V.M.; Nunes, S.P.; Peinemann, K.V. Self-Assembled Asymmetric Block Copolymer Membranes: Bridging the Gap from Ultra- to Nanofiltration. Angew. Chem. Int. Ed. 2015, 54, 13937–13941.
- Radjabian, M.; Abetz, V. Advanced porous polymer membranes from self-assembling block copolymers. Prog. Polym. Sci. 2020, 102, 101219.
- Cheng, X.Q.; Wang, Z.X.; Jiang, X.; Li, T.X.; Lau, C.H.; Guo, Z.H.; Ma, J.; Shao, L. Towards sustainable ultrafast molecular-separation membranes: From conventional polymers to emerging materials. Prog. Mater. Sci. 2018, 92, 258–283.
- Park, H.B.; Kamcev, J.; Robeson, L.M.; Elimelech, M.; Freeman, B.D. Maximizing the right stuff: The trade-off between membrane permeability and selectivity. Science 2017, 356, eaab0530.
- Khan, I.; Hou, F.J.; Le, H.P. The impact of natural resources, energy consumption, and population growth on environmental quality: Fresh evidence from the United States of America. Sci. Total Environ. 2021, 754, 142222.
- Schiermeier, Q. Increased flood risk linked to global warming. Nature 2011, 470, 316.
- Zandalinas, S.I.; Fritschi, F.B.; Mittler, R. Global Warming, Climate Change, and Environmental Pollution: Recipe for a Multifactorial Stress Combination Disaster. Trends Plant Sci. 2021, 26, 588–599.
- Fan, E.S.; Li, L.; Wang, Z.P.; Lin, J.; Huang, Y.X.; Yao, Y.; Chen, R.J.; Wu, F. Sustainable Recycling Technology for Li-Ion Batteries and Beyond: Challenges and Future Prospects. Chem. Rev. 2020, 120, 7020–7063.
- Orilall, M.C.; Wiesner, U. Block copolymer based composition and morphology control in nanostructured hybrid materials for energy conversion and storage: Solar cells, batteries, and fuel cells. Chem. Soc. Rev. 2011, 40, 520–535.
- Bates, F.S. Polymer-polymer phase behavior. Sci.-New York 1991, 251, 898–905.
- Wong, C.K.; Qiang, X.L.; Muller, A.H.E.; Groschel, A.H. Self-Assembly of block copolymers into internally ordered microparticles. Prog. Polym. Sci. 2020, 102, 101211.
- Feng, H.B.; Lu, X.Y.; Wang, W.Y.; Kang, N.G.; Mays, J.W. Block Copolymers: Synthesis, Self-Assembly, and Applications. Polymers 2017, 9, 494.
- Farrell, R.A.; Fitzgerald, T.G.; Borah, D.; Holmes, J.D.; Morris, M.A. Chemical Interactions and Their Role in the Microphase Separation of Block Copolymer Thin Films. Int. J. Mol. Sci. 2009, 10, 3671–3712.
- Bates, C.M.; Seshimo, T.; Maher, M.J.; Durand, W.J.; Cushen, J.D.; Dean, L.M.; Blachut, G.; Ellison, C.J.; Willson, C.G. Polarity-Switching Top Coats Enable Orientation of Sub-10-nm Block Copolymer Domains. Science 2012, 338, 775–779.
- Hagita, K.; Aoyagi, T.; Abe, Y.; Genda, S.; Honda, T. Deep learning-based estimation of Flory–Huggins parameter of A–B block copolymers from cross-sectional images of phase-separated structures. Sci. Rep. 2021, 11, 12322.
- Yoshida, H.; Takenaka, M. 1-Physics of block copolymers from bulk to thin films. In Directed Self-Assembly of Block Co-Polymers for Nano-Manufacturing; Gronheid, R., Nealey, P., Eds.; Woodhead Publishing: Sawston, UK, 2015; pp. 3–26.
- Liu, M.J.; Qiang, Y.C.; Li, W.H.; Qiu, F.; Shi, A.C. Stabilizing the Frank-Kasper Phases via Binary Blends of AB Diblock Copolymers. Acs Macro Lett. 2016, 5, 1167–1171.
- Bates, F.S.; Fredrickson, G.H. Block copolymers-Designer soft materials. Phys. Today 1999, 52, 32–38.
- Groschel, A.H.; Muller, A.H.E. Self-assembly concepts for multicompartment nanostructures. Nanoscale 2015, 7, 11841–11876.
- Wanka, G.; Hoffmann, H.; Ulbricht, W. Phase-Diagrams and aggregation behavior of poly(oxyethylene)-poly(oxypropylene)-poly(oxyethylene) triblock copolymers in aqueous-solutions. Macromolecules 1994, 27, 4145–4159.
- Bates, F.S.; Hillmyer, M.A.; Lodge, T.P.; Bates, C.M.; Delaney, K.T.; Fredrickson, G.H. Multiblock Polymers: Panacea or Pandora’s Box? Science 2012, 336, 434–440.
- Majewski, P.W.; Yager, K.G. Rapid ordering of block copolymer thin films. J. Phys.-Condes. Matter 2016, 28, 403002.
- Abetz, V.; Kremer, K.; Muller, M.; Reiter, G. Functional Macromolecular Systems: Kinetic Pathways to Obtain Tailored Structures. Macromol. Chem. Phys. 2019, 220, 1800334.
- Cui, H.G.; Chen, Z.Y.; Zhong, S.; Wooley, K.L.; Pochan, D.J. Block copolymer assembly via kinetic control. Science 2007, 317, 647–650.
- Lynd, N.A.; Meuler, A.J.; Hillmyer, M.A. Polydispersity and block copolymer self-assembly. Prog. Polym. Sci. 2008, 33, 875–893.
- Hadziioannou, G.; Skoulios, A. Molecular weight dependence of lamellar structure in styrene isoprene two- and three-block copolymers. Macromolecules 1982, 15, 258–262.
- Carrot, C.; Guillet, J. From dynamic moduli to molecular weight distribution: A study of various polydisperse linear polymers. J. Rheol. 1997, 41, 1203–1220.
- Llorens, J.; Rude, E.; Marcos, R.M. Polydispersity index from linear viscoelastic data: Unimodal and bimodal linear polymer melts. Polymer 2003, 44, 1741–1750.
- Levy, M. The impact of the concept of “Living Polymers” on material science. Polym. Adv. Technol. 2007, 18, 681–684.
- Nguyen, H.T.; Tran, T.T.; Nguyen-Thai, N.U. Preparation of polydisperse polystyrene-block-poly(4-vinyl pyridine) synthesized by TEMPO-mediated radical polymerization and the facile nanostructure formation by self-assembly. J. Nanostruct. Chem. 2018, 8, 61–69.
- Ruzette, A.V.; Tence-Girault, S.; Leibler, L.; Chauvin, F.; Bertin, D.; Guerret, O.; Gerard, P. Molecular disorder and mesoscopic order in polydisperse acrylic block copolymers prepared by controlled radical polymerization. Macromolecules 2006, 39, 5804–5814.
- Lynd, N.A.; Hillmyer, M.A. Effects of polydispersity on the order-disorder transition in block copolymer melts. Macromolecules 2007, 40, 8050–8055.
- Widin, J.M.; Schmitt, A.K.; Schmitt, A.L.; Im, K.; Mahanthappa, M.K. Unexpected Consequences of Block Polydispersity on the Self-Assembly of ABA Triblock Copolymers. J. Am. Chem. Soc. 2012, 134, 3834–3844.
- Hanley, K.J.; Lodge, T.P.; Huang, C.-I. Phase Behavior of a Block Copolymer in Solvents of Varying Selectivity. Macromolecules 2000, 33, 5918–5931.
- Wang, Y. Nondestructive Creation of Ordered Nanopores by Selective Swelling of Block Copolymers: Toward Homoporous Membranes. Acc. Chem. Res. 2016, 49, 1401–1408.
- Yi, Z.; Zhang, P.-B.; Liu, C.-J.; Zhu, L.-P. Symmetrical Permeable Membranes Consisting of Overlapped Block Copolymer Cylindrical Micelles for Nanoparticle Size Fractionation. Macromolecules 2016, 49, 3343–3351.
- Gohil, J.M.; Choudhury, R.R. Chapter 2-Introduction to Nanostructured and Nano-enhanced Polymeric Membranes: Preparation, Function, and Application for Water Purification. In Nanoscale Materials in Water Purification; Thomas, S., Pasquini, D., Leu, S.-Y., Gopakumar, D.A., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 25–57.
- Jiang, H.; Chen, T.; Chen, Z.; Huo, J.; Zhang, L.; Zhou, J. Computer simulations on double hydrophobic PS-b-PMMA porous membrane by non-solvent induced phase separation. Fluid Phase Equilib. 2020, 523, 112784.
- Paradiso, S.P.; Delaney, K.T.; Garcia-Cervera, C.J.; Ceniceros, H.D.; Fredrickson, G.H. Block Copolymer Self Assembly during Rapid Solvent Evaporation: Insights into Cylinder Growth and Stability. Acs Macro Lett. 2014, 3, 16–20.
- Russell, T.P.; Coulon, G.; Deline, V.R.; Miller, D.C. Characteristics of the surface-induced orientation for symmetric diblock PS/PMMA copolymers. Macromolecules 1989, 22, 4600–4606.
- Phillip, W.A.; O’Neill, B.; Rodwogin, M.; Hillmyer, M.A.; Cussler, E.L. Self-Assembled Block Copolymer Thin Films as Water Filtration Membranes. ACS Appl. Mater. Interfaces 2010, 2, 847–853.
- Tseng, Y.H.; Lin, Y.L.; Ho, J.H.; Chang, C.T.; Fan, Y.C.; Shen, M.H.; Chen, J.T. Reversible and tunable morphologies of amphiphilic block copolymer nanorods confined in nanopores: Roles of annealing solvents. Polymer 2021, 228, 123859.
- Lee, S.; Cheng, L.-C.; Gadelrab, K.R.; Ntetsikas, K.; Moschovas, D.; Yager, K.G.; Avgeropoulos, A.; Alexander-Katz, A.; Ross, C.A. Double-Layer Morphologies from a Silicon-Containing ABA Triblock Copolymer. ACS Nano 2018, 12, 6193–6202.
- Chavis, M.A.; Smilgies, D.M.; Wiesner, U.B.; Ober, C.K. Widely Tunable Morphologies in Block Copolymer Thin Films Through Solvent Vapor Annealing Using Mixtures of Selective Solvents. Adv. Funct. Mater. 2015, 25, 3057–3065.
- Peng, J.; Han, Y.C.; Knoll, W.; Kim, D.H. Development of nanodomain and fractal morphologies in solvent annealed block copolymer thin films. Macromol. Rapid Commun. 2007, 28, 1422–1428.
- Stenbock-Fermor, A.; Rudov, A.A.; Gumerov, R.A.; Tsarkova, L.A.; Boker, A.; Moller, M.; Potemkin, I.I. Morphology-Controlled Kinetics of Solvent Uptake by Block Copolymer Films in Nonselective Solvent Vapors. Acs Macro Lett. 2014, 3, 803–807.
- Tsarkova, L. Distortion of a Unit Cell versus Phase Transition to Nonbulk Morphology in Frustrated Films of Cylinder-Forming Polystyrene-b-polybutadiene Diblock Copolymers. Macromolecules 2012, 45, 7985–7994.
- Cavicchi, K.A.; Berthiaume, K.J.; Russell, T.P. Solvent annealing thin films of poly(isoprene-b-lactide). Polymer 2005, 46, 11635–11639.
- Xuan, Y.; Peng, J.; Cui, L.; Wang, H.; Li, B.; Han, Y. Morphology Development of Ultrathin Symmetric Diblock Copolymer Film via Solvent Vapor Treatment. Macromolecules 2004, 37, 7301–7307.
- Phillip, W.A.; Hillmyer, M.A.; Cussler, E.L. Cylinder Orientation Mechanism in Block Copolymer Thin Films Upon Solvent Evaporation. Macromolecules 2010, 43, 7763–7770.
- Lin, Z.Q.; Kim, D.H.; Wu, X.D.; Boosahda, L.; Stone, D.; LaRose, L.; Russell, T.P. A rapid route to arrays of nanostructures in thin films. Adv. Mater. 2002, 14, 1373–1376.
- Kim, S.H.; Misner, M.J.; Xu, T.; Kimura, M.; Russell, T.P. Highly oriented and ordered arrays from block copolymers via solvent evaporation. Adv. Mater. 2004, 16, 226–231.
- Kim, K.; Park, S.; Kim, Y.; Bang, J.; Park, C.; Ryu, D.Y. Optimized Solvent Vapor Annealing for Long-Range Perpendicular Lamellae in PS-b-PMMA Films. Macromolecules 2016, 49, 1722–1730.
- Li, X.; Peng, J.; Wen, Y.; Kim, D.H.; Knoll, W. Morphology change of asymmetric diblock copolymer micellar films during solvent annealing. Polymer 2007, 48, 2434–2443.
- Park, S.; Kim, Y.; Lee, W.; Hur, S.-M.; Ryu, D.Y. Gyroid Structures in Solvent Annealed PS-b-PMMA Films: Controlled Orientation by Substrate Interactions. Macromolecules 2017, 50, 5033–5041.
- Cheng, X.; Boker, A.; Tsarkova, L. Temperature-Controlled Solvent Vapor Annealing of Thin Block Copolymer Films. Polymers 2019, 11, 1312.
- Gowd, E.B.; Koga, T.; Endoh, M.K.; Kumar, K.; Stamm, M. Pathways of cylindrical orientations in PS-b-P4VP diblock copolymer thin films upon solvent vapor annealing. Soft Matter 2014, 10, 7753–7761.
- Bosworth, J.K.; Paik, M.Y.; Ruiz, R.; Schwartz, E.L.; Huang, J.Q.; Ko, A.W.; Smilgies, D.M.; Black, C.T.; Ober, C.K. Control of self-assembly of lithographically patternable block copolymer films. Acs Nano 2008, 2, 1396–1402.
- Arias-Zapata, J.; Bohme, S.; Garnier, J.; Girardot, C.; Legrain, A.; Zelsmann, M. Ultrafast Assembly of PS-PDMS Block Copolymers on 300 mm Wafers by Blending with Plasticizers. Adv. Funct. Mater. 2016, 26, 5690–5700.
- Guillen, G.R.; Ramon, G.Z.; Kavehpour, H.P.; Kaner, R.B.; Hoek, E.M.V. Direct microscopic observation of membrane formation by nonsolvent induced phase separation. J. Membr. Sci. 2013, 431, 212–220.
- Wang, D.-M.; Lai, J.-Y. Recent advances in preparation and morphology control of polymeric membranes formed by nonsolvent induced phase separation. Curr. Opin. Chem. Eng. 2013, 2, 229–237.
- Guillen, G.R.; Pan, Y.; Li, M.; Hoek, E.M.V. Preparation and Characterization of Membranes Formed by Nonsolvent Induced Phase Separation: A Review. Ind. Eng. Chem. Res. 2011, 50, 3798–3817.
- Tan, X.; Li, K. Inorganic hollow fibre membranes in catalytic processing. Curr. Opin. Chem. Eng. 2011, 1, 69–76.
- Plisko, T.V.; Penkova, A.V.; Burts, K.S.; Bildyukevich, A.V.; Dmitrenko, M.E.; Melnikova, G.B.; Atta, R.R.; Mazur, A.S.; Zolotarev, A.A.; Missyul, A.B. Effect of Pluronic F127 on porous and dense membrane structure formation via non-solvent induced and evaporation induced phase separation. J. Membr. Sci. 2019, 580, 336–349.
- Gu, Y.; Wiesner, U. Tailoring Pore Size of Graded Mesoporous Block Copolymer Membranes: Moving from Ultrafiltration toward Nanofiltration. Macromolecules 2015, 48, 6153–6159.
- Abed, M.R.M.; Kumbharkar, S.C.; Groth, A.M.; Li, K. Ultrafiltration PVDF hollow fibre membranes with interconnected bicontinuous structures produced via a single-step phase inversion technique. J. Membr. Sci. 2012, 407–408, 145–154.
- Gin, D.L.; Noble, R.D. Designing the Next Generation of Chemical Separation Membranes. Science 2011, 332, 674–676.
- Li, D.F.; Chung, T.S.; Ren, J.Z.; Wang, R. Thickness dependence of macrovoid evolution in wet phase-inversion asymmetric membranes. Ind. Eng. Chem. Res. 2004, 43, 1553–1556.
- Widjojo, N.; Chung, T.S. Thickness and air gap dependence of macrovoid evolution in phase-inversion asymmetric hollow fiber membranes. Ind. Eng. Chem. Res. 2006, 45, 7618–7626.
- Abetz, V. Isoporous Block Copolymer Membranes. Macromol. Rapid Commun. 2015, 36, 10–22.
- Foroutani, K.; Ghasemi, S.M.; Pourabbas, B. Molecular tailoring of polystyrene-block-poly (acrylic acid) block copolymer toward additive-free asymmetric isoporous membranes via SNIPS. J. Membr. Sci. 2021, 623, 119099.
- Peinemann, K.V.; Abetz, V.; Simon, P.F.W. Asymmetric superstructure formed in a block copolymer via phase separation. Nat. Mater. 2007, 6, 992–996.
- Karunakaran, M.; Nunes, S.P.; Qiu, X.Y.; Yu, H.Z.; Peinemann, K.V. Isoporous PS-b-PEO ultrafiltration membranes via self-assembly and water-induced phase separation. J. Membr. Sci. 2014, 453, 471–477.
- Stegelmeier, C.; Filiz, V.; Abetz, V.; Perlich, J.; Fery, A.; Ruckdeschel, P.; Rosenfeldt, S.; Forster, S. Topological Paths and Transient Morphologies during Formation of Mesoporous Block Copolymer Membranes. Macromolecules 2014, 47, 5566–5577.
- Jung, A.; Rangou, S.; Abetz, C.; Filiz, V.; Abetz, V. Structure Formation of Integral Asymmetric Composite Membranes of Polystyrene-block-Poly(2-vinylpyridine) on a Nonwoven. Macromol. Mater. Eng. 2012, 297, 790–798.
- Rahman, M.M. Selective Swelling and Functionalization of Integral Asymmetric Isoporous Block Copolymer Membranes. Macromol. Rapid Commun. 2021, 42, 2100235.
- Dorin, R.M.; Phillip, W.A.; Sai, H.; Werner, J.; Elimelech, M.; Wiesner, U. Designing block copolymer architectures for targeted membrane performance. Polymer 2014, 55, 347–353.
- Rangou, S.; Buhr, K.; Filiz, V.; Clodt, J.I.; Lademann, B.; Hahn, J.; Jung, A.; Abetz, V. Self-organized isoporous membranes with tailored pore sizes. J. Membr. Sci. 2014, 451, 266–275.
- Radjabian, M.; Abetz, C.; Fischer, B.; Meyer, A.; Abetz, V. Influence of Solvent on the Structure of an Amphiphilic Block Copolymer in Solution and in Formation of an Integral Asymmetric Membrane. Acs Appl. Mater. Interfaces 2017, 9, 31224–31234.
- Tan, K.W.; Jung, B.; Werner, J.G.; Rhoades, E.R.; Thompson, M.O.; Wiesner, U. Transient laser heating induced hierarchical porous structures from block copolymer-directed self-assembly. Science 2015, 349, 54–58.
- Seshimo, T.; Maeda, R.; Odashima, R.; Takenaka, Y.; Kawana, D.; Ohmori, K.; Hayakawa, T. Perpendicularly oriented sub-10-nm block copolymer lamellae by atmospheric thermal annealing for one minute. Sci. Rep. 2016, 6, 19481.
- Albalak, R.J.; Thomas, E.L.; Capel, M.S. Thermal annealing of roll-cast triblock copolymer films. Polymer 1997, 38, 3819–3825.
- Tong, Q.Q.; Zheng, Q.; Sibener, S.J. Alignment and Structural Evolution of Cylinder-Forming Diblock Copolymer Thin Films in Patterned Tapered-Width Nanochannels. Macromolecules 2014, 47, 4236–4242.
- Sepe, A.; Hoppe, E.T.; Jaksch, S.; Magerl, D.; Zhong, Q.; Perlich, J.; Posselt, D.; Smilgies, D.M.; Papadakis, C.M. The effect of heat treatment on the internal structure of nanostructured block copolymer films. J. Phys.-Condes. Matter 2011, 23, 254213.
- Shi, L.-Y.; Yin, C.; Zhou, B.; Xia, W.; Weng, L.; Ross, C.A. Annealing Process Dependence of the Self-Assembly of Rod–Coil Block Copolymer Thin Films. Macromolecules 2021, 54, 1657–1664.
- Majewski, P.W.; Yager, K.G. Latent Alignment in Pathway-Dependent Ordering of Block Copolymer Thin Films. Nano Lett. 2015, 15, 5221–5228.
- Wang, H.S.; Kim, K.H.; Bang, J. Thermal Approaches to Perpendicular Block Copolymer Microdomains in Thin Films: A Review and Appraisal. Macromol. Rapid Commun. 2019, 40, 1800728.
- Stehlin, F.; Diot, F.; Gwiazda, A.; Dirani, A.; Salaun, M.; Zelsmann, M.; Soppera, O. Local Reorganization of Diblock Copolymer Domains in Directed Self-Assembly Monitored by in Situ High-Temperature AFM. Langmuir 2013, 29, 12796–12803.
- Majewski, P.W.; Yager, K.G. Reordering transitions during annealing of block copolymer cylinder phases. Soft Matter 2016, 12, 281–294.
- Zhou, Z.P.; Liu, G.L. Controlling the Pore Size of Mesoporous Carbon Thin Films through Thermal and Solvent Annealing. Small 2017, 13, 1603107.
- Kim, S.; Jeon, G.; Heo, S.W.; Kim, H.J.; Kim, S.B.; Chang, T.; Kim, J.K. High aspect ratio cylindrical microdomains oriented vertically on the substrate using block copolymer micelles and temperature-programmed solvent vapor annealing. Soft Matter 2013, 9, 5550–5556.
- Kim, E.; Ahn, H.; Park, S.; Lee, H.; Lee, M.; Lee, S.; Kim, T.; Kwak, E.-A.; Lee, J.H.; Lei, X.; et al. Directed Assembly of High Molecular Weight Block Copolymers: Highly Ordered Line Patterns of Perpendicularly Oriented Lamellae with Large Periods. ACS Nano 2013, 7, 1952–1960.
- Gurovich, E. On Microphase Separation of Block Copolymers In an Electric Field: Four Universal Classes. Macromolecules 1994, 27, 7339–7362.
- Boker, A.; Elbs, H.; Hansel, H.; Knoll, A.; Ludwigs, S.; Zettl, H.; Urban, V.; Abetz, V.; Muller, A.H.E.; Krausch, G. Microscopic mechanisms of electric-field-induced alignment of block copolymer microdomains. Phys. Rev. Lett. 2002, 89, 135502.
- Schmidt, K.; Schoberth, H.G.; Ruppel, M.; Zettl, H.; Hansel, H.; Weiss, T.M.; Urban, V.; Krausch, G.; Boker, A. Reversible tuning of a block-copolymer nanostructure via electric fields. Nat. Mater. 2008, 7, 142–145.
- Schoberth, H.G.; Olszowka, V.; Schmidt, K.; Boker, A. Effects of Electric Fields on Block Copolymer Nanostructures. In Complex Macromolecular Systems I; Muller, A.H.E., Schmidt, H.W., Eds.; Springer: Amsterdam, The Netherlands, 2010; Volume 227, pp. 1–31.
- Olszowka, V.; Hund, M.; Kuntermann, V.; Scherdel, S.; Tsarkova, L.; Boker, A. Electric Field Alignment of a Block Copolymer Nanopattern: Direct Observation of the Microscopic Mechanism. Acs Nano 2009, 3, 1091–1096.
- Schoberth, H.G.; Pester, C.W.; Ruppe, M.; Urban, V.S.; Boker, A. Orientation-Dependent Order-Disorder Transition of Block Copolymer Lamellae in Electric Fields. Acs Macro Lett. 2013, 2, 469–473.
- Marencic, A.P.; Chaikin, P.M.; Register, R.A. Orientational order in cylinder-forming block copolymer thin films. Phys. Rev. E 2012, 86, 021507.
- Morrison, F.; Le Bourvellec, G.; Winter, H.H. Flow-induced structure and rheology of a triblock copolymer. J. Appl. Polym. Sci. 1987, 33, 1585–1600.
- Angelescu, D.E.; Waller, J.H.; Adamson, D.H.; Deshpande, P.; Chou, S.Y.; Register, R.A.; Chaikin, P.M. Macroscopic orientation of block copolymer cylinders in single-layer films by shearing. Adv. Mater. 2004, 16, 1736–1740.
- Angelescu, D.E.; Waller, J.H.; Register, R.A.; Chaikin, P.M. Shear-induced alignment in thin films of spherical nanodomains. Adv. Mater. 2005, 17, 1878–1881.
- Pujari, S.; Keaton, M.A.; Chaikin, P.M.; Register, R.A. Alignment of perpendicular lamellae in block copolymer thin films by shearing. Soft Matter 2012, 8, 5358–5363.
- Davis, R.L.; Chaikin, P.M.; Register, R.A. Cylinder Orientation and Shear Alignment in Thin Films of Polystyrene-Poly(n-hexyl methacrylate) Diblock Copolymers. Macromolecules 2014, 47, 5277–5285.
- Marencic, A.P.; Adamson, D.H.; Chaikin, P.M.; Register, R.A. Shear alignment and realignment of sphere-forming and cylinder-forming block-copolymer thin films. Phys. Rev. E 2010, 81, 011503.
- Kim, S.Y.; Nunns, A.; Gwyther, J.; Davis, R.L.; Manners, I.; Chaikin, P.M.; Register, R.A. Large-Area Nanosquare Arrays from Shear-Aligned Block Copolymer Thin Films. Nano Lett. 2014, 14, 5698–5705.
- Wang, X.J.; Goswami, M.; Kumar, R.; Sumpter, B.G.; Mays, J. Morphologies of block copolymers composed of charged and neutral blocks. Soft Matter 2012, 8, 3036–3052.
- Sing, C.E.; Zwanikken, J.W.; de la Cruz, M.O. Electrostatic control of block copolymer morphology. Nat. Mater. 2014, 13, 694–698.
- Goswami, M.; Sumpter, B.G.; Mays, J. Controllable stacked disk morphologies of charged diblock copolymers. Chem. Phys. Lett. 2010, 487, 272–278.
- Zhou, H.; Liu, C.G.; Gao, C.Q.; Qu, Y.Q.; Shi, K.Y.; Zhang, W.Q. Polymerization-Induced Self-Assembly of Block Copolymer Through Dispersion RAFT Polymerization in Ionic Liquid. J. Polym. Sci. Pol. Chem. 2016, 54, 1517–1525.
- Kim, S.Y.; Yoon, E.; Joo, T.; Park, M.J. Morphology and Conductivity in Ionic Liquid Incorporated Sulfonated Block Copolymers. Macromolecules 2011, 44, 5289–5298.
- Bennett, T.M.; Jack, K.S.; Thurecht, K.J.; Blakey, I. Perturbation of the Experimental Phase Diagram of a Diblock Copolymer by Blending with an Ionic Liquid. Macromolecules 2016, 49, 205–214.
- Cui, T.; Li, X.; Dong, B.; Li, X.; Guo, M.; Wu, L.; Li, B.; Li, H. Janus onions of block copolymers via confined self-assembly. Polymer 2019, 174, 70–76.
- He, Q.B.; Zhang, Y.J.; Li, H.L.; Chen, Q. Rheological Properties of ABA-Type Copolymers Physically End-Cross-Linked by Polyoxometalate. Macromolecules 2020, 53, 10927–10941.
- Zhang, L.Y.; Liu, C.; Shang, H.Y.; Cao, X.; Chai, S.C.; Chen, Q.; Wu, L.X.; Li, H.L. Electrostatic tuning of block copolymer morphologies by inorganic macroions. Polymer 2016, 106, 53–61.
More
Information
Subjects:
Nanoscience & Nanotechnology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.5K
Revisions:
2 times
(View History)
Update Date:
15 Nov 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No