Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Yan Yik Lim | -- | 1690 | 2022-11-08 03:11:19 | | | |
| 2 | Beatrix Zheng | + 1 word(s) | 1691 | 2022-11-10 02:08:21 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Lim, Y.Y.; Zaidi, A.M.A.; Miskon, A. Drug Carriers for Arthritis Therapy. Encyclopedia. Available online: https://encyclopedia.pub/entry/33598 (accessed on 10 August 2026).
Lim YY, Zaidi AMA, Miskon A. Drug Carriers for Arthritis Therapy. Encyclopedia. Available at: https://encyclopedia.pub/entry/33598. Accessed August 10, 2026.
Lim, Yan Yik, Ahmad Mujahid Ahmad Zaidi, Azizi Miskon. "Drug Carriers for Arthritis Therapy" Encyclopedia, https://encyclopedia.pub/entry/33598 (accessed August 10, 2026).
Lim, Y.Y., Zaidi, A.M.A., & Miskon, A. (2022, November 09). Drug Carriers for Arthritis Therapy. In Encyclopedia. https://encyclopedia.pub/entry/33598
Lim, Yan Yik, et al. "Drug Carriers for Arthritis Therapy." Encyclopedia. Web. 09 November, 2022.
Copy Citation
Drug carriers are used to penetrate the biological barriers and deliver the requisite active pharmaceutical ingredients (API). This has greater significance for medication in arthritis therapies because the inflammatory progression of rheumatoid arthritis (RA) and osteoarthritis (OA) is complex, intertwined, and influenced by one another. For this purpose, the drug delivery systems (DDS) of this carrier are currently being investigated with exogenous triggers and endogenous stimuli. This flexible-release invention will solve the problems of multiple implantations and local toxic accumulation at the lesion sites.
exogenous triggers
endogenous stimuli
drug delivery systems
1. Introduction
Programmable arthritis therapy [1] is in high demand now because the current therapy still lacks efficacy [2] and no recovery therapy has been discovered. Arthritis therapy is complex, intertwined, and progressively influenced by rheumatoid arthritis (RA) and osteoarthritis (OA) [3]. The most difficult part is the progression of pathological inflammation from OA to RA, which involves mediators, such as synovial hyperplasia and pannus synovitis [4]. Generally, RA and OA are chronic and joint-degenerative diseases, respectively, that are classified as inflammatory arthritis (IA) [5] and non-IA [6], respectively. Commonly, OA and RA are specified for large joints of the hip and knee, and small joints of the hands and wrists, such as metatarsophalangeal and interphalangeal toe joints, respectively [7]. The symptoms of OA begin with joint pain, cartilage degeneration, and osseous overgrowth, whereas RA inherits the OA characteristics and then progresses to cartilage destruction and function loss [7]. These severely restrict the patients’ mobility and activities, resulting in disability and living quality deterioration, respectively.
Although many approaches, such as drug administration routes [8], intra-articular therapies [9], resections [10], reconstruction surgeries [11], and articular cartilage scaffolds [12] , have been undertaken, the problems of drug delivery systems (DDS), such as mediate infiltration, pharmacokinetics, pharmacodynamics, efficacy, systemic toxicity, and navigation control, still remain unsatisfactory. Moreover, arthritis therapy involves multiple inflammatory factors, resulting in difficult medication [13]. Therefore, the current therapy is limited to alleviating these disease symptoms [14] or pathological alterations. Nonetheless, they have the same pathological microenvironmental changes that could be used as alterations to design biosensors in DDS [15]. These unique alteration features could be characterised as biosensors [16] in scaffold structures [17] , which act as detectors to be sensitively cleaved by the selective endogenous stimuli for active pharmaceutical ingredient (API) releases [18]. As a result, an alternative tissue engineering approach with a better sustained-release feature in DDS holds tremendous promise in target-specified delivery, on-demand stimulation, on-program triggering, and great flexibility [19]. This should be urgently developed towards achieving therapy efficacy and patient compliance in clinical translations [20].
2. Status Quo of Drug Carriers for Arthritis Therapy
The drug carrier for arthritis therapy is progressing from traditional to systemic drug administration routes [8]. The traditional drugs are non-steroidal anti-inflammatory drugs (NSAID) and glucocorticoids, which are commonly used in traditional drug administration routes, such as oral, topical, transdermal, and injection [21]. The systemic drug administration routes, such as intra-articular, surgery, and drug carriers, use API such as disease-modifying anti-rheumatic drugs (DMARD), disease-modifying OA drugs (DMOAD), and biological response regulators [22]. However, the traditional intra-articular therapies, resection, and reconstruction surgeries are still unsatisfactory due to the infiltration, efficacy, drug clearance, and target localisation ability. In particular are the radio-, chemo-, immune-, and hormone therapies after osteosarcoma and tumour surgeries, which may result in systemic side effects [23] such as secondary injury, increased risk of recurrence, and immunological rejection [24][25]. Over a long period of therapy, these may increase the detoxification burdens of the liver, kidney, and bone marrow system, resulting from the toxic doses accumulated [26]. Due to these numerous potential systemic side-effects, it is a complication of therapeutic efficacy that limits their clinical applications. Therefore, a new class of drug carriers with better DDS is continuously being created.
2.1. Historical Development
The historical development of DDS is highlighted with a timeline of their significant discoveries and advancements, as shown in Figure 1. The first drug carrier as hydrogel, mostly used in DDS, was invented by Wichterle and Lím in 1960 via a 3D cross-linked polymer, as shown in the green background in Figure 1 [27]. This hydrogel has become one of the most popular drug carriers because of its nanosize particles, API affinity-based nature, bioactive release control, and injectable designs. The first injectable hydrogel using hyaluronic acid (HA) was invented by Rydell and Balazs in 1971 [28]. Later, HA was most commonly used in pain relief for OA therapy. Until 1980, the hydrogel was used as DDS, becoming popular after a drug-release device with rate-controlling was invented by Lee et al. [29]. Even though many thermal therapies were used before 1986, the thermal stimulus was first introduced by Helme et al., which was used to release immune-reactive substance P for inflammation therapies, as shown in the orange background in Figure 1 [30]. In 1987, the enzymatic stimulus using antigen-induced arthritis was first introduced by Bonanomi et al. to investigate the pharmacokinetics and therapeutic effects [31]. In 1998, the pH stimulus using pH-sensitive P2X receptors [32] was first introduced by Dowd et al. to study peripheral nociceptive afferents in the rat knee joint [33]. P2X receptors are the membrane ion channels that open in response to the binding of extracellular adenosine 5′-triphosphate (ATP). In 2000, the redox stimulus using Cu/Zn-superoxide dismutase (SOD) loaded with Fc receptor was first introduced by Vouldoukis et al. to study apoptosis [34]. In 2014, the electro-ion stimulus using electrostatic interaction between negatively charged proteoglycans and highly positively charged Avidin was first introduced by Bajpayee et al. to investigate its pharmacodynamics effects [35].
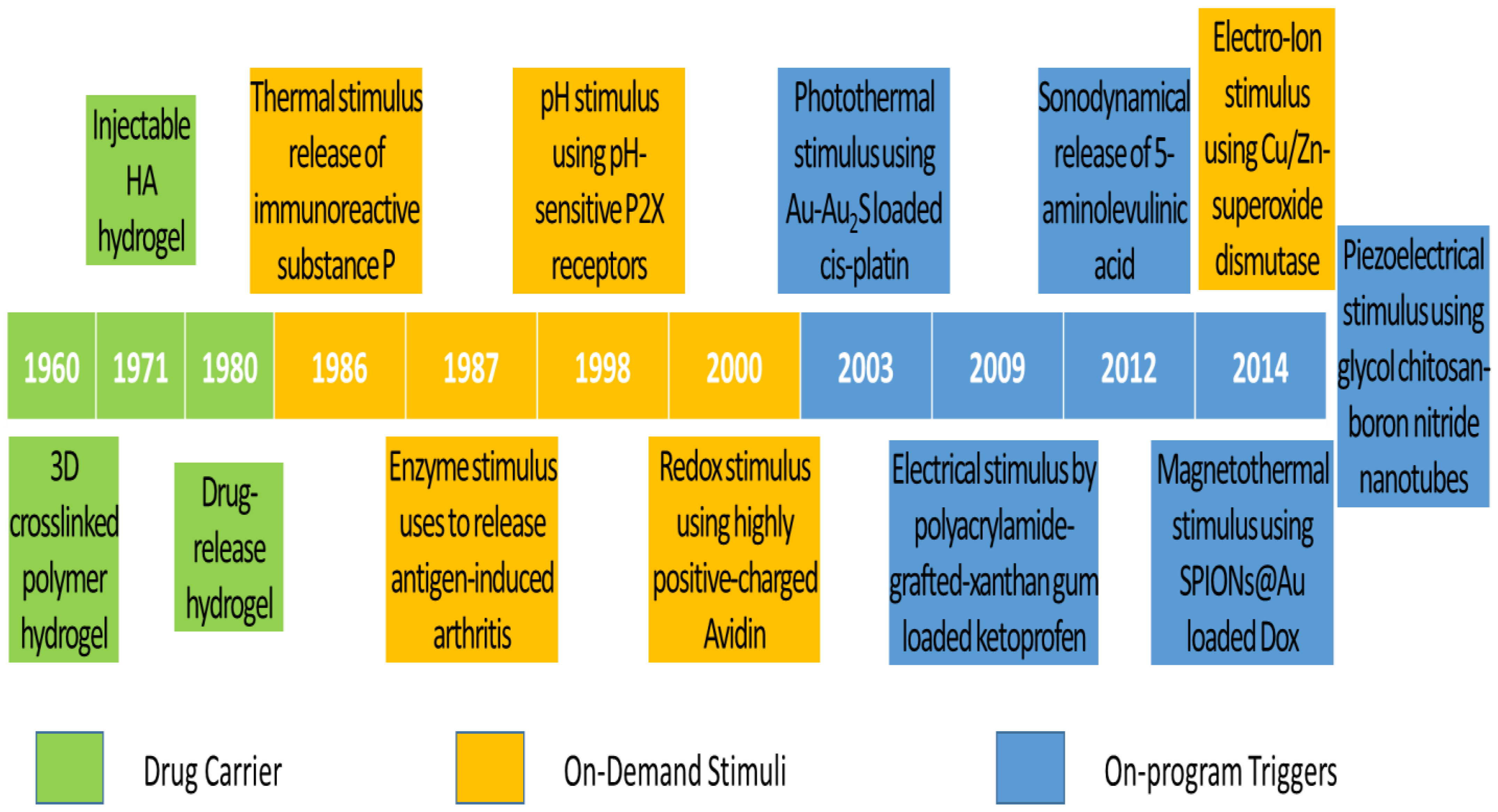
Figure 1. Timeline of significant discoveries and advancements in drug delivery systems with drug carriers, on-demand stimuli, and on-program triggers.
The existing DDS still does not meet the demands of scientists and researchers. Therefore, more flexible drug release systems or those composed of the existing trigger systems are being developed. Ren and Chow (2003) used a photothermal trigger with Au-Au2S-loaded cis-platin for near infra-red (NIR) DDS, as shown in the blue background in Figure 1 [36]. This DDS was a composed stimulation of photo and thermal, where gold linkages were leaked by NIR to free cis-platin for thermal stimulation. In 2009, Kulkarni and Sa used an electrical trigger with polyacrylamide-grafted xanthan-gum-loaded ketoprofen for transdermal DDS [37]. It was a simple study of in vitro drug release influenced by an electrical trigger. In addition, in 2012, Lv et al. used a sonodynamical trigger of 5-aminolevulinic acid therapy to investigate anti-carcinoma efficiency [38]. In these in vivo and in vitro studies using low-intensity US vibration, they found efficacy for SAS cell line proliferation and apoptosis. However, it is regretful that the increment of ROS in the therapy process was not further investigated. In 2014, Mohammad and Yusof developed a magnetothermal trigger using gold-coated superparamagnetic iron oxide nanoparticles (SPIONs@Au) loaded with doxorubicin (Dox) as an anthracycline-type chemotherapy drug for cancer therapy [39]. This in vitro DDS found that the cell viability and proliferation were more efficacious under low frequency oscillatory magnetic stimulation, but there was no effect under thermal stimulation. In the same year, Ricotti et al. used a piezoelectrical trigger of glycol chitosan-boron nitride nanotubes with US transducer stimulation for modulation of F/G-actin [40]. This simulation used a US transducer to convert mechanical stress into electrical signals to investigate actin expression in human dermal fibroblasts. From these highlights of the timeline figure, the on-program trigger began to synergistically correspond to the variations of on-demand stimuli to improve therapeutic efficacy. As a result of this work in the past 10 years, these multiple-stimulus trigger DDS strategies have received dramatic progression due to rapid advanced bio-fabrication technologies.
2.2. Clinical Advancement
In the last 10 years, many novel drug carriers have been developed, but there has not been much completed clinical testing. Thus, there are 10 drug carriers for arthritis-related therapies that have completed the recruitment status of the United States Federal Government clinical trials identifiers (GCTI) [41]. There are four, two, two, one, and one drug carriers successfully invented for knee OA [42], RA [43], osteosarcoma [44], joint infections [45], and peri-implant bone therapies [46], respectively. For the primary tests, the researchers observed that there were 5 pharmacokinetics tests out of 10 [47]. Moreover, the Western Ontario and McMaster Universities OA Index (WOMAC) is used as a self-administered health status for non-IA patients, such as knee OA patients, for pain management [48]. These dominant pharmacokinetics tests mean that the target concentration, distribution volume, and elimination clearance of API are the main considerations of clinical trials [49]. As a result, drug carriers with efficacy, programming release, and patient compliance features are the most preferable choices for scientists.
As shown in Table 1, drug carriers are generally classified into targeted biological-disease-modifying products and products resulting from the conjugation of hydrophilic and hydrophobic functional groups. The microRNA (miR)-146a [50] drug carrier is the targeted biological-disease-modifying product used to release tocilizumab [51][52]. MiR exhibits neurological disease dysfunction in the different stages of the central nervous system, which is not related to this topic and not further elaborated in this research. The other nine drug carriers are created from both functional group conjugations, such as multi-layer lipid in lyophilised formulation (TLC599), diclofenac etalhyaluronate (SI-613), corticosteroid fluticasone propionate with polyvinyl alcohol (PVA) coating (EP-104IAR), HA polynucleotides (PN) [53], tumour necrosis factor (TNF) inhibitor (adalimumab) [54][55], albumin-bound inhibitor with mammalian target of rapamycin (mTOR) (ABI-009) [56], sirolimus derivatives with mTOR inhibitor kinase (temsirolimus), active Ted compound in phosphate ester prodrug (tedizolid phosphate) [57], and platelet-rich fibrin (PRF) [58]. The hydrophilic functional groups are designed to carry API, such as dexamethasone (Dex) sodium phosphate [59], diclofenac [60][61], fluticasone propionate [62], PN [63][64], methotrexate (Mtx) [65][66], nivolumab [67][68], liposomal Dox [69][70], tedizolid (Ted) [71][72], and simvastatin [73][74], which cleave autonomously with endogenous stimuli. However, these inventions still have not solved the systemic toxicity problem due to their long-term uses. Therefore, more advanced inventions with on-program triggers are moving into clinical trials. As a result, this research elaborates on the inventions resulting from the composition of on-demand stimuli and on-program triggers.
Table 1. Drug carriers for arthritis therapy, API, GCTI and year completed, and primary tests.
| Drug Carrier | Arthritis Therapy | API | GCT I, Year | Primary Test | Refs. |
|---|---|---|---|---|---|
| TLC599 (BioSeizer®) | Knee OA | Dex sodium phosphate | NCT03754049, 2022 | Pharmacokinetics | [75][76] |
| SI-613 | Knee OA | Diclofenac (Voltaren®) | NCT03209362, 2021 | WOMAC | [77][78] |
| EP-104IAR | Knee OA | Fluticasone propionate | NCT02609126, 2021 | Pharmacokinetics | [79][80] |
| HA-PN (Condrotide® Plus) | Knee OA | PN | NCT02417610, 2017 | WOMAC | [81][82] |
| miR-146a | RA | Tocilizumab (Actemra®) | NCT03149796, 2017 | miR expressions | [83][84] |
| Adalimumab (Humira®) | RA | Mtx | NCT01185288, 2014 | Pharmacokinetics | [85][86] |
| ABI-009 | Osteosarcoma | Nivolumab (Opdivo®) | NCT03190174, 2021 | Pharmacokinetics | [87][88] |
| Temsirolimus (Torisel®) | Osteosarcoma | Liposomal Dox (Doxil®) | NCT00949325, 2019 | Pharmacokinetics | [89][90] |
| Ted phosphate (Sivextro®) | Joint infections | Ted | NCT03378427, 2021 | Immuno-compromised | [91][92] |
| PRF | Peri-implant bone | Simvastatin (Zocor®) | NCT05008068, 2021 | Bone regeneration | [93][94] |
References
- Wu, C.; Cheng, J.; Li, W.; Yang, L.; Dong, H.; Zhang, X. Programmable Polymeric Microneedles for Combined Chemotherapy and Antioxidative Treatment of Rheumatoid Arthritis. ACS Appl. Mater. Interfaces 2021, 13, 55559–55568.
- Lorenzo, H.K.; Susin, S.A. Therapeutic potential of AIF-mediated caspase-independent programmed cell death. Drug Resist. Updates 2007, 10, 235–255.
- Michallek, F.; Ulas, S.T.; Poddubnyy, D.; Proft, F.; Schneider, U.; Hermann, K.-G.A.; Dewey, M.; Diekhoff, T. Fractal analysis of perfusion imaging in synovitis: A novel imaging biomarker for grading inflammatory activity based on assessing angiogenesis. RMD Open 2022, 8, e002078.
- Zhang, B.; Xiao, L.; Zhou, H.; Li, M.; Wang, J.; Guo, L. Application of Dynamic Contrast-Enhanced MRI in the Diagnosis of Rheumatoid Arthritis. Contrast Media Mol. Imaging 2022, 2022, 3055465.
- Medrado, L.N.; Mendonça, M.L.M.; Budib, M.B.; Oliveira-Junior, S.A.; Martinez, P.F. Effectiveness of aquatic exercise in the treatment of inflammatory arthritis: Systematic review. Rheumatol. Int. 2022, 42, 1681–1691.
- Peshkova, M.; Lychagin, A.; Lipina, M.; Di Matteo, B.; Anzillotti, G.; Ronzoni, F.; Kosheleva, N.; Shpichka, A.; Royuk, V.; Fomin, V.; et al. Gender-Related Aspects in Osteoarthritis Development and Progression: A Review. Int. J. Mol. Sci. 2022, 23, 2767.
- Kean, W.F.; Kean, R.; Buchanan, W.W. Osteoarthritis: Symptoms, signs and source of pain. Inflammopharmacology 2004, 12, 3–31.
- Li, Z.A.; Sant, S.; Cho, S.K.; Goodman, S.B.; Bunnell, B.A.; Tuan, R.S.; Gold, M.S.; Lin, H. Synovial joint-on-a-chip for modeling arthritis: Progress, pitfalls, and potential. Trends Biotechnol. 2022.
- de la Torre-Aboki, J.; Uson, J.; Pitsillidou, I.; Vardanyan, V.; Nikiphorou, E.; Rodriguez-Garcia, S.C.; Castellanos-Moreira, R.; Pandit, H.; O’Neill, T.W.; Doherty, M.; et al. Intra-articular therapies: Patient preferences and professional practices in European countries. Rheumatol. Int. 2022, 42, 869–878.
- Yang, Q.; Lu, W.; You, T.; Zhang, X.; Zhang, W.; Li, C. Effectiveness of arthroscopic distal clavicle resection for symptomatic acromioclavicular joint arthritis. Chin. J. Reparative Reconstr. Surg. 2022, 36, 698–702.
- Wolford, L.M.; Kesterke, M.J. Does Combined Temporomandibular Joint Reconstruction With Patient-Fitted Total Joint Prosthesis and Orthognathic Surgery Provide Stable Skeletal and Occlusal Outcomes in Juvenile Idiopathic Arthritis Patients? J. Oral Maxillofac. Surg. 2022, 80, 138–150.
- Lim, Y.Y.; Miskon, A.; Zaidi, A.M.A. Structural Strength Analyses for Low Brass Filler Biomaterial with Anti-Trauma Effects in Articular Cartilage Scaffold Design. Materials 2022, 15, 4446.
- Paul, A.K.; Jahan, R.; Paul, A.; Mahboob, T.; Bondhon, T.A.; Jannat, K.; Hasan, A.; Nissapatorn, V.; Wilairatana, P.; de Lourdes Pereira, M.; et al. The Role of Medicinal and Aromatic Plants against Obesity and Arthritis: A Review. Nutrients 2022, 14, 985.
- Yang, J.; Xiong, J.; Guo, Y.; Gu, L.; Wu, P.; Tong, M.; Liu, C.; Sun, J. B7-H3 blockade decreases macrophage inflammatory response and alleviates clinical symptoms of arthritis. Immunol. Lett. 2022, 242, 46–53.
- Li, C.; Zheng, X.; Hu, M.; Jia, M.; Jin, R.; Nie, Y. Recent progress in therapeutic strategies and biomimetic nanomedicines for rheumatoid arthritis treatment. Expert Opin. Drug Deliv. 2022, 19, 883–898.
- Cicha, I.; Priefer, R.; Severino, P.; Souto, E.B.; Jain, S. Biosensor-Integrated Drug Delivery Systems as New Materials for Biomedical Applications. Biomolecules 2022, 12, 1198.
- Lim, Y.Y.; Miskon, A.; Zaidi, A.M.A.; Megat Ahmad, M.M.H.; Abu Bakar, M. Structural Characterization Analyses of Low Brass Filler Biomaterial for Hard Tissue Implanted Scaffold Applications. Materials 2022, 15, 1421.
- Verdes, M.; Mace, K.; Margetts, L.; Cartmell, S. Status and challenges of electrical stimulation use in chronic wound healing. Curr. Opin. Biotechnol. 2022, 75, 102710.
- Popa, M.; Atanase, L.I. Biological macromolecules for drug delivery in tissue engineering. In Biological Macromolecules; Elsevier: Amsterdam, The Netherlands, 2022; pp. 393–418.
- Seppen, B.; Wiegel, J.; Ter Wee, M.; Schaardenburg, D.; Roorda, L.; Nurmohamed, M.; Boers, M.; Bos, W. Smartphone-Assisted Patient-Initiated Care Versus Usual care in Patients With Rheumatoid Arthritis and Low Disease Activity: A Randomized Controlled Trial. Arthritis Rheumatol. 2022, 1–9.
- Ding, Q.; Shiltz, D.; Hossami, D.; Konieczny, A.M. The economic burden of biologic disease-modifying antirheumatic drugs in rheumatoid arthritis patients in the United States. Expert Rev. Pharm. Outcomes Res. 2022, 22, 1–11.
- Kim, M.; Choe, Y.; Lee, S. Lessons From the Success and Failure of Targeted Drugs for Rheumatoid Arthritis: Perspectives for Effective Basic and Translational Research. Immune Netw. 2022, 22, e8.
- Hussain, N.; Brull, R.; Speer, J.; Hu, L.-Q.; Sawyer, T.; McCartney, C.J.L.; Abdallah, F.W. Analgesic benefits of the quadratus lumborum block in total hip arthroplasty: A systematic review and meta-analysis. Anaesthesia 2022, 77, 1152–1162.
- Ilfeld, B.M.; Plunkett, A.; Vijjeswarapu, A.M.; Hackworth, R.; Dhanjal, S.; Turan, A.; Cohen, S.P.; Eisenach, J.C.; Griffith, S.; Hanling, S.; et al. Percutaneous Neuromodulation of the Brachial Plexus and Sciatic Nerve for the Treatment of Acute Pain Following Surgery: Secondary Outcomes From a Multicenter, Randomized, Controlled Pilot Study. Neuromodul. Technol. Neural Interface 2022.
- Gómez-Puerta, J.A.; Lobo-Prat, D.; Perez-García, C.; Ponce, A.; Frade-sosa, B.; Millán Arciniegas, A.M.; Ojeda, F.; Ruiz-Esquide, V.; Corominas, H. Clinical Patterns and Follow-Up of Inflammatory Arthritis and Other Immune-Related Adverse Events Induced by Checkpoint Inhibitors. A Multicenter Study. Front. Med. 2022, 9, 888377.
- Ostoich, P.; Beltcheva, M.; Antonio Heredia Rojas, J.; Metcheva, R. Radionuclide Contamination as a Risk Factor in Terrestrial Ecosystems: Occurrence, Biological Risk, and Strategies for Remediation and Detoxification. In The Toxicity of Environmental Pollutants; IntechOpen: London, UK, 2022.
- Wichterle, O.; Lím, D. Hydrophilic Gels for Biological Use. Nature 1960, 185, 117–118.
- Rydell, N.M.D.; Balazs, E.A.M.D. Effect of Intra-articular Injection of Hyaluronic Acid on the Clinical Symptoms of Osteoarthritis and on Granulation Tissue Formation. Clin. Orthop. Relat. Res. 1971, 80, 25–32.
- Lee, E.S.; Kim, S.W.; Kim, S.H.; Cardinal, J.R.; Jacobs, H. Drug release from hydrogel devices with ratecontrolling barriers. J. Membr. Sci. 1980, 7, 293–303.
- Helme, R.D.; Koschorke, G.M.; Zimmermann, M. Immunoreactive substance P release from skin nerves in the rat by noxious thermal stimulation. Neurosci. Lett. 1986, 63, 295–299.
- Bonanomi, M.H.; Velvart, M.; Stimpel, M.; Roos, K.M.; Fehr, K.; Weder, H.G. Studies of pharmacokinetics and therapeutic effects of glucocorticoids entrapped in liposomes after intraarticular application in healthy rabbits and in rabbits with antigen-induced arthritis. Rheumatol. Int. 1987, 7, 203–212.
- North, R.A. Molecular Physiology of P2X Receptors. Physiol. Rev. 2002, 82, 1013–1067.
- Dowd, E.; McQueen, D.S.; Chessell, I.P.; Humphrey, P.P.A. P2X receptor-mediated excitation of nociceptive afferents in the normal and arthritic rat knee joint. Br. J. Pharmacol. 1998, 125, 341–346.
- Vouldoukis, I.; Sivan, V.; Vozenin, M.C.; Kamaté, C.; Calenda, A.; Mazier, D.; Dugas, B. Fc-Receptor-Mediated Intracellular Delivery of Cu/Zn-superoxide Dismutase (SOD1) Protects Against Redox-Induced Apoptosis Through a Nitric Oxide Dependent Mechanism. Mol. Med. 2000, 6, 1042–1053.
- Bajpayee, A.G.; Wong, C.R.; Bawendi, M.G.; Frank, E.H.; Grodzinsky, A.J. Avidin as a model for charge driven transport into cartilage and drug delivery for treating early stage post-traumatic osteoarthritis. Biomaterials 2014, 35, 538–549.
- Ren, L.; Chow, G.M. Synthesis of nir-sensitive Au–Au2S nanocolloids for drug delivery. Mater. Sci. Eng. C 2003, 23, 113–116.
- Kulkarni, R.V.; Sa, B. Electroresponsive Polyacrylamide-grafted-xanthan Hydrogels for Drug Delivery. J. Bioact. Compat. Polym. 2009, 24, 368–384.
- Lv, Y.; Fang, M.; Zheng, J.; Yang, B.; Li, H.; Xiuzigao, Z.; Song, W.; Chen, Y.; Cao, W. Low-intensity Ultrasound Combined with 5-aminolevulinic Acid Administration in the Treatment of Human Tongue Squamous Carcinoma. Cell. Physiol. Biochem. 2012, 30, 321–333.
- Mohammad, F.; Yusof, N.A. Doxorubicin-loaded magnetic gold nanoshells for a combination therapy of hyperthermia and drug delivery. J. Colloid Interface Sci. 2014, 434, 89–97.
- Ricotti, L.; das Neves, R.P.; Ciofani, G.; Canale, C.; Nitti, S.; Mattoli, V.; Mazzolai, B.; Ferreira, L.; Menciassi, A. Boron nitride nanotube-mediated stimulation modulates F/G-actin ratio and mechanical properties of human dermal fibroblasts. J. Nanopart. Res. 2014, 16, 2247.
- Ehrhardt, S.; Appel, L.J.; Meinert, C.L. Trends in National Institutes of Health Funding for Clinical Trials Registered in ClinicalTrials.gov. JAMA 2015, 314, 2566.
- Pereira, T.V.; Jüni, P.; Saadat, P.; Xing, D.; Yao, L.; Bobos, P.; Agarwal, A.; Hincapié, C.A.; da Costa, B.R. Viscosupplementation for knee osteoarthritis: Systematic review and meta-analysis. BMJ 2022, 157, e069722.
- Hurysz, B.; Bottini, N. Emerging proteoglycans and proteoglycan-targeted therapies in rheumatoid arthritis. Am. J. Physiol.-Cell Physiol. 2022, 322, C1061–C1067.
- Moukengue, B.; Lallier, M.; Marchandet, L.; Baud’huin, M.; Verrecchia, F.; Ory, B.; Lamoureux, F. Origin and Therapies of Osteosarcoma. Cancers 2022, 14, 3503.
- Le Vavasseur, B.; Zeller, V. Antibiotic Therapy for Prosthetic Joint Infections: An Overview. Antibiotics 2022, 11, 486.
- Albrektsson, T.; Tengvall, P.; Amengual-Peñafiel, L.; Coli, P.; Kotsakis, G.; Cochran, D.L. Implications of considering peri-implant bone loss a disease, a narrative review. Clin. Implant. Dent. Relat. Res. 2022, 24, 532–543.
- Pantaleão, S.Q.; Fernandes, P.O.; Gonçalves, J.E.; Maltarollo, V.G.; Honorio, K.M. Recent Advances in the Prediction of Pharmacokinetics Properties in Drug Design Studies: A Review. ChemMedChem 2022, 17, 1–13.
- Held, M.B.; Gazgalis, A.; Neuwirth, A.L.; Shah, R.P.; Cooper, H.J.; Geller, J.A. Imageless robotic-assisted total knee arthroplasty leads to similar 24-month WOMAC scores as compared to conventional total knee arthroplasty: A retrospective cohort study. Knee Surg. Sport. Traumatol. Arthrosc. 2022, 30, 2631–2638.
- Atkinson, A.J. Clinical pharmacokinetics. In Atkinson’s Principles of Clinical Pharmacology; Elsevier: Amsterdam, The Netherlands, 2022; pp. 11–26.
- Fan, W.; Liang, C.; Ou, M.; Zou, T.; Sun, F.; Zhou, H.; Cui, L. MicroRNA-146a Is a Wide-Reaching Neuroinflammatory Regulator and Potential Treatment Target in Neurological Diseases. Front. Mol. Neurosci. 2020, 13, 90.
- Rivellese, F.; Surace, A.E.A.; Goldmann, K.; Sciacca, E.; Çubuk, C.; Giorli, G.; John, C.R.; Nerviani, A.; Fossati-Jimack, L.; Thorborn, G.; et al. Rituximab versus tocilizumab in rheumatoid arthritis: Synovial biopsy-based biomarker analysis of the phase 4 R4RA randomized trial. Nat. Med. 2022, 28, 1256–1268.
- Huang, J.; Chen, Z.; Zhao, L.; Cheng, Y.; Gao, M.; Huang, C.; Wang, Z.; Wang, F. Tocilizumab in rheumatoid arthritis-associated peripheral ulcerative keratitis: A 1-year follow-up case report. Rheumatol. Autoimmun. 2022, 2, 45–50.
- Lee, Y.J.; Kim, H.T.; Lee, Y.J.; Paik, S.H.; Moon, Y.S.; Lee, W.J.; Chang, S.E.; Lee, M.W.; Choi, J.H.; Jung, J.M.; et al. Comparison of the effects of polynucleotide and hyaluronic acid fillers on periocular rejuvenation: A randomized, double-blind, split-face trial. J. Dermatol. Treat. 2022, 33, 254–260.
- Lu, X.; Hu, R.; Peng, L.; Liu, M.; Sun, Z. Efficacy and Safety of Adalimumab Biosimilars: Current Critical Clinical Data in Rheumatoid Arthritis. Front. Immunol. 2021, 12, 638444.
- Raza, N.; Nair, N.; Plant, D.; Hyrich, K.; Morgan, A.W.; Isaacs, J.; Wilson, A.G.; Barton, A. P189 A longitudinal study of psychological predictors of response to adalimumab in patients with rheumatoid arthritis. Rheumatology 2022, 61, keac133.188.
- Wagner, A.J.; Ravi, V.; Ganjoo, K.N.; Van Tine, B.A.; Riedel, R.F.; Chugh, R.; Cranmer, L.D.; Gordon, E.M.; Hornick, J.L.; Kwiatkowski, D.J.; et al. ABI-009 (nab-sirolimus) in advanced malignant perivascular epithelioid cell tumors (PEComa): Preliminary efficacy, safety, and mutational status from AMPECT, an open label phase II registration trial. J. Clin. Oncol. 2019, 37 (Suppl. S15), 11005.
- Fala, L. Sivextro (Tedizolid Phosphate) Approved for the Treatment of Adults with Acute Bacterial Skin and Skin-Structure Infections. Am. Health Drug Benefits 2015, 8, 111–115.
- Tavakoli, M.; Mirhaj, M.; Labbaf, S.; Varshosaz, J.; Taymori, S.; Jafarpour, F.; Salehi, S.; Abadi, S.A.M.; Sepyani, A. Fabrication and evaluation of Cs/PVP sponge containing platelet-rich fibrin as a wound healing accelerator: An in vitro and in vivo study. Int. J. Biol. Macromol. 2022, 204, 245–257.
- Li, M.; Vora, L.K.; Peng, K.; Donnelly, R.F. Trilayer microneedle array assisted transdermal and intradermal delivery of dexamethasone. Int. J. Pharm. 2022, 612, 121295.
- Kubo, T.; Kumai, T.; Ikegami, H.; Kano, K.; Nishii, M.; Seo, T. Diclofenac–hyaluronate conjugate (diclofenac etalhyaluronate) intra-articular injection for hip, ankle, shoulder, and elbow osteoarthritis: A randomized controlled trial. BMC Musculoskelet. Disord. 2022, 23, 371.
- Branco, A.C.; Oliveira, A.S.; Monteiro, I.; Nolasco, P.; Silva, D.C.; Figueiredo-Pina, C.G.; Colaço, R.; Serro, A.P. PVA-Based Hydrogels Loaded with Diclofenac for Cartilage Replacement. Gels 2022, 8, 143.
- Kumar, V.; Sethi, B.; Yanez, E.; Leung, D.H.; Ghanwatkar, Y.Y.; Cheong, J.; Tso, J.; Narang, A.S.; Nagapudi, K.; Mahato, R.I. Effect of magnesium stearate surface coating method on the aerosol performance and permeability of micronized fluticasone propionate. Int. J. Pharm. 2022, 615, 121470.
- Jang, J.Y.; Kim, J.H.; Kim, M.W.; Kim, S.H.; Yong, S.Y. Study of the Efficacy of Artificial Intelligence Algorithm-Based Analysis of the Functional and Anatomical Improvement in Polynucleotide Treatment in Knee Osteoarthritis Patients: A Prospective Case Series. J. Clin. Med. 2022, 11, 2845.
- Hummer, C.D.; Huang, Y.; Sheehan, B. Adherence to the OARSI recommendations for designing, conducting, and reporting of clinical trials in knee osteoarthritis: A targeted literature review. BMC Musculoskelet. Disord. 2022, 23, 171.
- García-González, C.M.; Baker, J. Treatment of early rheumatoid arthritis: Methotrexate and beyond. Curr. Opin. Pharmacol. 2022, 64, 102227.
- Giollo, A.; Fuzzi, E.; Doria, A. Methotrexate in early rheumatoid arthritis: Is the anchor drug still holding? Autoimmun. Rev. 2022, 21, 103031.
- Doki, Y.; Ajani, J.A.; Kato, K.; Xu, J.; Wyrwicz, L.; Motoyama, S.; Ogata, T.; Kawakami, H.; Hsu, C.-H.; Adenis, A.; et al. Nivolumab Combination Therapy in Advanced Esophageal Squamous-Cell Carcinoma. N. Engl. J. Med. 2022, 386, 449–462.
- Mittal, N.; Seyboth, B.; Kent, P. Checkpoint inhibitor and multireceptor tyrosine kinase inhibitor combination in relapsed refractory sarcomas: A single institution series. J. Clin. Oncol. 2022, 40 (Suppl. S16), 11529.
- Wen, X.; Pan, Q.; Xu, B.; Xiao, W.; Weng, D.; Zhao, J.; Xu, H.; Huang, Z.; Niu, X.; Zhang, X. Phase I study of pegylated liposomal doxorubicin and cisplatin in patients with advanced osteosarcoma. Cancer Chemother. Pharmacol. 2022, 89, 209–215.
- Huang, G.; Hua, S.; Liu, H.; Zhou, H.; Chen, X.; Wang, Z.; Yu, W. Efficacy of ifosfamide combined with liposome doxorubicin on osteosarcoma and its effects on serum IL-10, TNF-α, and IFN-γ in patients with osteosarcoma. Am. J. Transl. Res. 2022, 14, 1288–1296.
- Iqbal, K.; Milioudi, A.; Wicha, S.G. Pharmacokinetics and Pharmacodynamics of Tedizolid. Clin. Pharmacokinet. 2022, 61, 489–503.
- Jorda, A.; Wulkersdorfer, B.; Schörgenhofer, C.; Matzneller, P.; Al Jalali, V.; Bauer, M.; Wölf-Duchek, M.; Lackner, E.; Dorn, C.; Jilma, B.; et al. Influence of tedizolid on the cytokine response to the endotoxin challenge in healthy volunteers: A cross-over trial. J. Antimicrob. Chemother. 2022, 77, 1424–1431.
- García-Sobrino, R.; Casado-Losada, I.; Bruno-Pérez, L.; García, C.; Reinecke, H.; Elvira, C.; Rodríguez-Hernández, J.; Gallardo, A.; Martínez-Campos, E. Thermosensitive hydrogels functionalized with pH sensitive COOH groups for bone cell harvesting. Eur. Polym. J. 2022, 169, 111131.
- Diniz, J.A.; Barbirato, D.D.S.; do Nascimento, E.H.L.; dos Anjos Pontual, A.; Dourado, A.C.A.G.; Laureano Filho, J.R. Tomographic evaluation of the effect of simvastatin topical use on alveolar bone microarchitecture, pain and swelling after mandibular third molar extraction: A randomized controlled trial. Clin. Oral Investig. 2022, 26, 3533–3545.
- Hunter, D.J.; Chang, C.-C.; Wei, J.C.-C.; Lin, H.-Y.; Brown, C.; Tai, T.-T.; Wu, C.-F.; Chuang, W.C.-M.; Shih, S.-F. TLC599 in patients with osteoarthritis of the knee: A phase IIa, randomized, placebo-controlled, dose-finding study. Arthritis Res. Ther. 2022, 24, 52.
- Brown, C.; Wu, C.-F.; Chuang, W.; Shih, S.-F. Single intra-articular injection of TLC599 in patients with osteoarthritis knee pain: Subgroup analyses of a placebo-controlled 24-week phase 2 trial. Osteoarthr. Cartil. 2020, 28, S481–S482.
- Kisukeda, T.; Onaya, J.; Yoshioka, K. Effect of diclofenac etalhyaluronate (SI-613) on the production of high molecular weight sodium hyaluronate in human synoviocytes. BMC Musculoskelet. Disord. 2019, 20, 201.
- Nishida, Y.; Kano, K.; Osato, T.; Seo, T. Open-label phase 3 study of diclofenac conjugated to hyaluronate (diclofenac etalhyaluronate: ONO-5704/SI-613) for treatment of osteoarthritis: 1-year follow-up. BMC Musculoskelet. Disord. 2021, 22, 233.
- Malone, A.; Price, J.; Price, N.; Peck, V.; Getgood, A.; Petrella, R.; Helliwell, J. Safety and pharmacokinetics of EP-104IAR (sustained-release fluticasone propionate) in knee osteoarthritis: A randomized, double-blind, placebo-controlled phase 1 trial. Osteoarthr. Cartil. Open 2021, 3, 100213.
- Getgood, A.; Dhollander, A.; Malone, A.; Price, J.; Helliwell, J. Pharmacokinetic Profile of Intra-articular Fluticasone Propionate Microparticles in Beagle Dog Knees. CARTILAGE 2017, 10, 139–147.
- Stagni, C.; Rocchi, M.; Mazzotta, A.; Del Piccolo, N.; Rani, N.; Govoni, M.; Vivarelli, L.; Veronesi, F.; Fini, M.; Dallari, D. Randomised, double-blind comparison of a fixed co-formulation of intra-articular polynucleotides and hyaluronic acid versus hyaluronic acid alone in the treatment of knee osteoarthritis: Two-year follow-up. BMC Musculoskelet. Disord. 2021, 22, 1–12.
- Kim, J.H.; Kwon, T.-R.; Lee, S.E.; Na Jang, Y.; Han, H.S.; Mun, S.K.; Kim, B.J. Comparative Evaluation of the Effectiveness of Novel Hyaluronic Acid-Polynucleotide Complex Dermal Filler. Sci. Rep. 2020, 10, 5127.
- Sabbatinelli, J.; Giuliani, A.; Matacchione, G.; Latini, S.; Laprovitera, N.; Pomponio, G.; Ferrarini, A.; Baroni, S.S.; Pavani, M.; Moretti, M.; et al. Decreased serum levels of the inflammaging marker miR-146a are associated with clinical non-response to tocilizumab in COVID-19 patients. Mech. Ageing Dev. 2020, 193, 111413.
- Zisman, D.; Safieh, M.; Simanovich, E.; Feld, J.; Kinarty, A.; Zisman, L.; Gazitt, T.; Haddad, A.; Elias, M.; Rosner, I.; et al. Tocilizumab (TCZ) Decreases Angiogenesis in Rheumatoid Arthritis Through Its Regulatory Effect on miR-146a-5p and EMMPRIN/CD147. Front. Immunol. 2021, 12, 739592.
- Coates, L.C.; Tillett, W.; D’Agostino, M.-A.; Rahman, P.; Behrens, F.; McDearmon-Blondell, E.L.; Bu, X.; Chen, L.; Kapoor, M.; Conaghan, P.G.; et al. Comparison between adalimumab introduction and methotrexate dose escalation in patients with inadequately controlled psoriatic arthritis (CONTROL): A randomised, open-label, two-part, phase 4 study. Lancet Rheumatol. 2022, 4, e262–e273.
- Burmester, G.-R.; Kivitz, A.J.; Kupper, H.; Arulmani, U.; Florentinus, S.; Goss, S.L.; Rathmann, S.S.; Fleischmann, R.M. Efficacy and safety of ascending methotrexate dose in combination with adalimumab: The randomised CONCERTO trial. Ann. Rheum. Dis. 2015, 74, 1037–1044.
- Gordon, E.M.; Chua-Alcala, V.S.; Kim, K.; Baby, R.; Angel, N.; Quon, D.; Wong, S.; Chawla, S.P. A phase I/II investigation of nivolumab and ABI-009 (nab-sirolimus) in advanced undifferentiated pleomorphic sarcoma (UPS), liposarcoma (LPS), chondrosarcoma (CS), osteosarcoma (OS), and Ewing sarcoma: Preliminary efficacy and safety results. J. Clin. Oncol. 2019, 37 (Suppl. S15), 11057.
- Hou, S.; Schmid, A.; Desai, N. Abstract 348: ABI-009 (nab-Sirolimus) improves tumor accumulation and antitumor activity over oral mTOR inhibitors. Exp. Mol. Ther. 2019, 79, 348.
- Huang, S.; Du, K.; Liu, Z.; Li, J. Inhibition of mTOR by temsirolimus overcomes radio-resistance in nasopharyngeal carcinoma. Clin. Exp. Pharmacol. Physiol. 2022, 49, 703–709.
- Laukkanen, S.; Veloso, A.B.; Yan, C.; Oksa, L.; Alpert, E.J.; Do, D.; Hyvärinen, N.; McCarthy, K.; Adhikari, A.; Yang, Q.; et al. Combination therapies to inhibit LCK tyrosine kinase and mTOR signaling in T-cell Acute Lymphoblastic Leukemia. Blood 2022, 140.
- Wong, E.; Rab, S. Tedizolid phosphate (sivextro): A second-generation oxazolidinone to treat acute bacterial skin and skin structure infections. Pharm. Ther. 2014, 39, 555–579.
- Vashistha, V.K.; Verma, N.; Kumar, R.; Tyagi, I.; Gaur, A.; Bala, R. Enantioseparation of linezolid and tedizolid using validated high-performance liquid chromatographic method. Chirality 2022, 34, 1044–1052.
- Raafat, S.N.; Amin, R.M.; Elmazar, M.M.; Khattab, M.M.; El-Khatib, A.S. The sole and combined effect of simvastatin and platelet rich fibrin as a filling material in induced bone defect in tibia of albino rats. Bone 2018, 117, 60–69.
- Gupta, S.; Verma, P.; Tikku, A.P.; Chandra, A.; Yadav, R.K.; Bharti, R.; Bains, R. “Effect of local application of simvastatin in bone regeneration of peri-apical defects-a clinico-radiographic study. J. Oral Biol. Craniofac. Res. 2020, 10, 583–591.
More
Information
Subjects:
Pharmacology & Pharmacy
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.4K
Entry Collection:
Biopharmaceuticals Technology
Revisions:
2 times
(View History)
Update Date:
10 Nov 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No