 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Rebecca Heinz | -- | 2458 | 2022-11-07 22:01:51 | | | |
| 2 | Amina Yu | + 13 word(s) | 2471 | 2022-11-08 02:33:47 | | | | |
| 3 | Amina Yu | -1 word(s) | 2470 | 2022-11-10 01:47:33 | | |
Video Upload Options
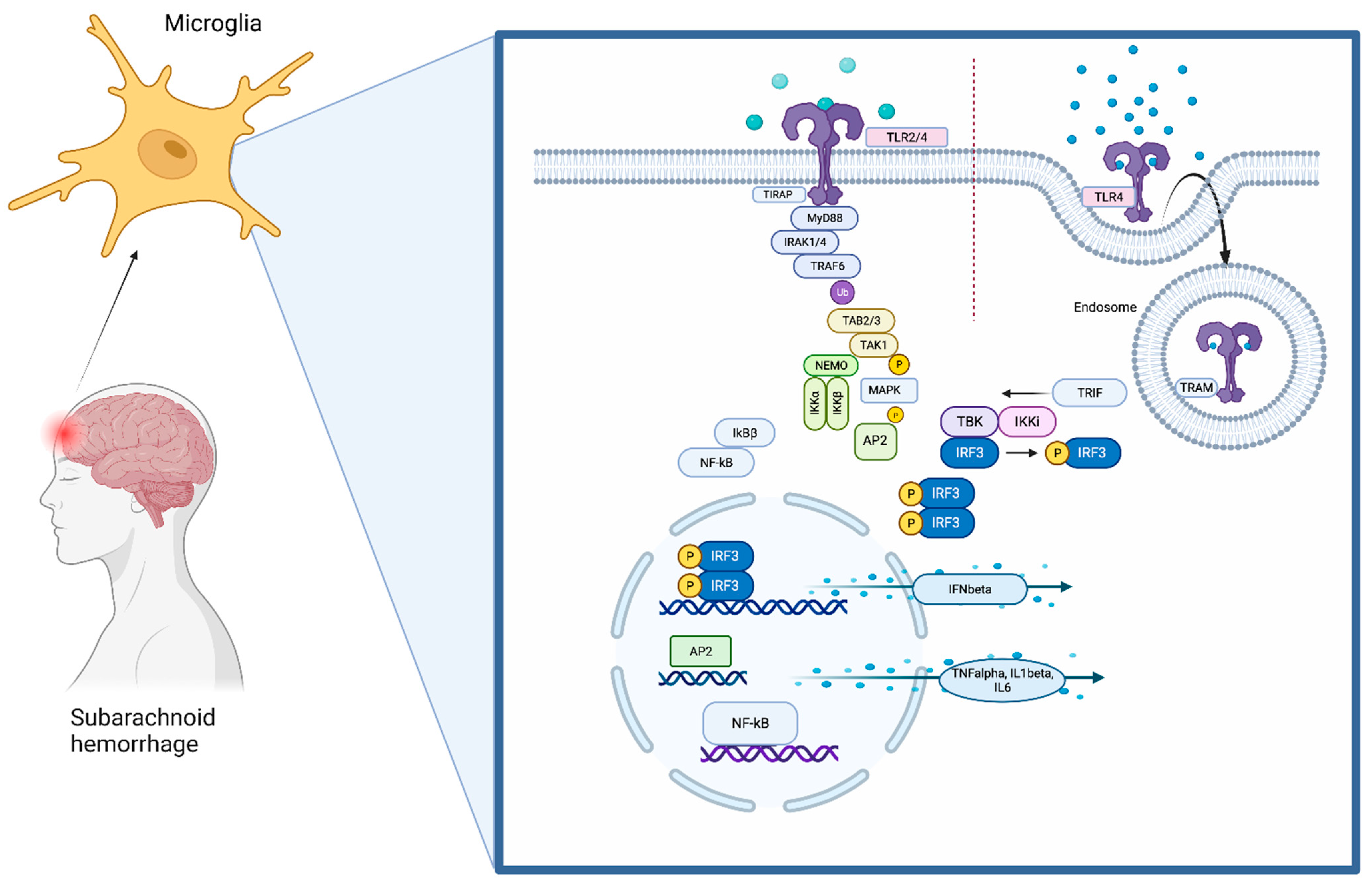
Subarachnoid hemorrhage (SAH) is a complex condition with high morbidity and mortality. Numerous different factors contributing to brain injury after SAH have been identified. Despite treatment of complications of early brain injury, such as rise of intracranial pressure, disturbance of the blood–brain barrier, cerebral edema, and decrease in cerebral perfusion, the outcome of many patients remains devastating. Neuroinflammation within the brain parenchyma with associated neuronal cell death has been described as a leading mechanism for additional secondary brain injury. It involves complex signaling cascades in which the upregulation of inflammatory genes is induced. Consequently, the release of different inflammatory cytokines and chemokines leads to self-reinforcement of the immune system with concomitant neuronal cell death, destroying the brain. The immune system within the central nervous system (CNS) fulfills a special role. It is privileged and differs in its standards from the peripheral immune response.
1. The Toll-Like Receptors (TLRs) Pathway
2. Crosstalk of TLR4 and the NLRP3 Inflammasome
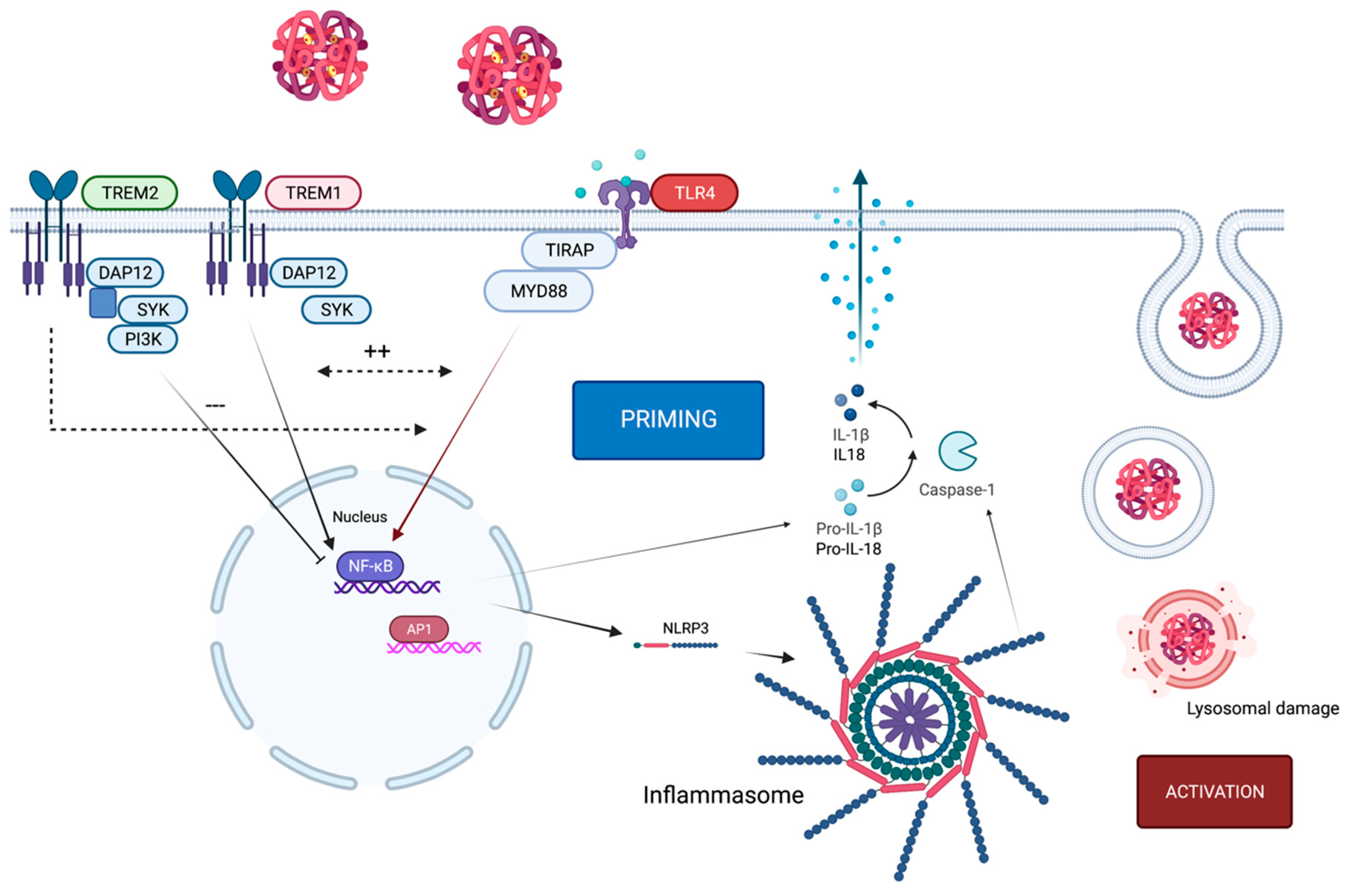
3. TLR4-Pathway-Associated Proteins Acting as Biomarkers following SAH
3.1. Ligands of TLR4
3.1.1. HMGB1
3.1.2. Red Blood Cell Lysate
3.2. Inflammatory Cytokines
3.3. TLR4
3.4. TREM1
3.5. Inflammasome Proteins
References
- Hanafy, K.A. The role of microglia and the TLR4 pathway in neuronal apoptosis and vasospasm after subarachnoid hemorrhage. J. Neuroinflammation 2013, 10, 83.
- Park, B.S.; Song, D.H.; Kim, H.M.; Choi, B.S.; Lee, H.; Lee, J.O. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature 2009, 458, 1191–1195.
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820.
- Yamamoto, M.; Sato, S.; Hemmi, H.; Hoshino, K.; Kaisho, T.; Sanjo, H.; Takeuchi, O.; Sugiyama, M.; Okabe, M.; Takeda, K.; et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science 2003, 301, 640–643.
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801.
- Palsson-McDermott, E.M.; Doyle, S.L.; McGettrick, A.F.; Hardy, M.; Husebye, H.; Banahan, K.; Gong, M.; Golenbock, D.; Espevik, T.; O’Neill, L.A. TAG, a splice variant of the adaptor TRAM, negatively regulates the adaptor MyD88-independent TLR4 pathway. Nat. Immunol. 2009, 10, 579–586.
- Tseng, P.H.; Matsuzawa, A.; Zhang, W.; Mino, T.; Vignali, D.A.; Karin, M. Different modes of ubiquitination of the adaptor TRAF3 selectively activate the expression of type I interferons and proinflammatory cytokines. Nat. Immunol. 2010, 11, 70–75.
- Hoebe, K.; Du, X.; Georgel, P.; Janssen, E.; Tabeta, K.; Kim, S.O.; Goode, J.; Lin, P.; Mann, N.; Mudd, S.; et al. Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature 2003, 424, 743–748.
- Huai, W.; Song, H.; Wang, L.; Li, B.; Zhao, J.; Han, L.; Gao, C.; Jiang, G.; Zhang, L.; Zhao, W. Phosphatase PTPN4 preferentially inhibits TRIF-dependent TLR4 pathway by dephosphorylating TRAM. J. Immunol. 2015, 194, 4458–4465.
- Liu, Y.; Dai, Y.; Li, Q.; Chen, C.; Chen, H.; Song, Y.; Hua, F.; Zhang, Z. Beta-amyloid activates NLRP3 inflammasome via TLR4 in mouse microglia. Neurosci. Lett. 2020, 736, 135279.
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832.
- Rathinam, V.A.; Vanaja, S.K.; Fitzgerald, K.A. Regulation of inflammasome signaling. Nat. Immunol. 2012, 13, 333–342.
- Walsh, J.G.; Muruve, D.A.; Power, C. Inflammasomes in the CNS. Nat. Rev. Neurosci. 2014, 15, 84–97.
- Xu, P.; Hong, Y.; Xie, Y.; Yuan, K.; Li, J.; Sun, R.; Zhang, X.; Shi, X.; Li, R.; Wu, J.; et al. TREM-1 Exacerbates Neuroinflammatory Injury via NLRP3 Inflammasome-Mediated Pyroptosis in Experimental Subarachnoid Hemorrhage. Transl. Stroke Res. 2020, 12, 643–659.
- He, Y.; Hara, H.; Nunez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 2016, 41, 1012–1021.
- Kwon, M.S.; Woo, S.K.; Kurland, D.B.; Yoon, S.H.; Palmer, A.F.; Banerjee, U.; Iqbal, S.; Ivanova, S.; Gerzanich, V.; Simard, J.M. Methemoglobin is an endogenous toll-like receptor 4 ligand-relevance to subarachnoid hemorrhage. Int. J. Mol. Sci. 2015, 16, 5028–5046.
- Nakahara, T.; Tsuruta, R.; Kaneko, T.; Yamashita, S.; Fujita, M.; Kasaoka, S.; Hashiguchi, T.; Suzuki, M.; Maruyama, I.; Maekawa, T. High-mobility group box 1 protein in CSF of patients with subarachnoid hemorrhage. Neurocrit. Care 2009, 11, 362–368.
- Zhong, W.J.; Duan, J.X.; Liu, T.; Yang, H.H.; Guan, X.X.; Zhang, C.Y.; Yang, J.T.; Xiong, J.B.; Zhou, Y.; Guan, C.X.; et al. Activation of NLRP3 inflammasome up-regulates TREM-1 expression in murine macrophages via HMGB1 and IL-18. Int. Immunopharmacol. 2020, 89 Pt A, 107045.
- Yang, J.; Wise, L.; Fukuchi, K.I. TLR4 Cross-Talk With NLRP3 Inflammasome and Complement Signaling Pathways in Alzheimer’s Disease. Front. Immunol. 2020, 11, 724.
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673.
- Guarda, G.; Braun, M.; Staehli, F.; Tardivel, A.; Mattmann, C.; Forster, I.; Farlik, M.; Decker, T.; Pasquier, R.A.D.; Romero, P.; et al. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity 2011, 34, 213–223.
- Zhang, X.; Wu, Q.; Zhang, Q.; Lu, Y.; Liu, J.; Li, W.; Lv, S.; Zhou, M.; Zhang, X.; Hang, C. Resveratrol Attenuates Early Brain Injury after Experimental Subarachnoid Hemorrhage via Inhibition of NLRP3 Inflammasome Activation. Front. Neurosci. 2017, 11, 611.
- Yang, S.J.; Shao, G.F.; Chen, J.L.; Gong, J. The NLRP3 Inflammasome: An Important Driver of Neuroinflammation in Hemorrhagic Stroke. Cell Mol. Neurobiol. 2018, 38, 595–603.
- Gao, L.; Dong, Q.; Song, Z.; Shen, F.; Shi, J.; Li, Y. NLRP3 inflammasome: A promising target in ischemic stroke. Inflamm. Res. 2017, 66, 17–24.
- Chen, G.Y.; Nunez, G. Sterile inflammation: Sensing and reacting to damage. Nat. Rev. Immunol. 2010, 10, 826–837.
- Hemmer, S.; Senger, S.; Griessenauer, C.J.; Simgen, A.; Oertel, J.; Geisel, J.; Hendrix, P. Admission serum high mobility group box 1 (HMGB1) protein predicts delayed cerebral ischemia following aneurysmal subarachnoid hemorrhage. Neurosurg. Rev. 2022, 45, 807–817.
- Harris, H.E.; Andersson, U.; Pisetsky, D.S. HMGB1: A multifunctional alarmin driving autoimmune and inflammatory disease. Nat. Rev. Rheumatol. 2012, 8, 195–202.
- Chaudhry, S.R.; Guresir, A.; Stoffel-Wagner, B.; Fimmers, R.; Kinfe, T.M.; Dietrich, D.; Lamprecht, A.; Vatter, H.; Guresir, E.; Muhammad, S. Systemic High-Mobility Group Box-1: A Novel Predictive Biomarker for Cerebral Vasospasm in Aneurysmal Subarachnoid Hemorrhage. Crit. Care Med. 2018, 46, e1023–e1028.
- Zhao, X.D.; Mao, H.Y.; Lv, J.; Lu, X.J. Expression of high-mobility group box-1 (HMGB1) in the basilar artery after experimental subarachnoid hemorrhage. J. Clin. Neurosci. 2016, 27, 161–165.
- Zhu, X.D.; Chen, J.S.; Zhou, F.; Liu, Q.C.; Chen, G.; Zhang, J.M. Relationship between plasma high mobility group box-1 protein levels and clinical outcomes of aneurysmal subarachnoid hemorrhage. J. Neuroinflammation 2012, 9, 194.
- Ieong, C.; Sun, H.; Wang, Q.; Ma, J. Glycyrrhizin suppresses the expressions of HMGB1 and ameliorates inflammative effect after acute subarachnoid hemorrhage in rat model. J. Clin. Neurosci. 2018, 47, 278–284.
- Haruma, J.; Teshigawara, K.; Hishikawa, T.; Wang, D.; Liu, K.; Wake, H.; Mori, S.; Takahashi, H.K.; Sugiu, K.; Date, I.; et al. Anti-high mobility group box-1 (HMGB1) antibody attenuates delayed cerebral vasospasm and brain injury after subarachnoid hemorrhage in rats. Sci. Rep. 2016, 6, 37755.
- Chaudhry, S.R.; Hafez, A.; Jahromi, B.R.; Kinfe, T.M.; Lamprecht, A.; Niemela, M.; Muhammad, S. Role of Damage Associated Molecular Pattern Molecules (DAMPs) in Aneurysmal Subarachnoid Hemorrhage (aSAH). Int. J. Mol. Sci. 2018, 19, 2035.
- Gram, M.; Sveinsdottir, S.; Ruscher, K.; Hansson, S.R.; Cinthio, M.; Akerstrom, B.; Ley, D. Hemoglobin induces inflammation after preterm intraventricular hemorrhage by methemoglobin formation. J. Neuroinflammation 2013, 10, 100.
- Wang, K.C.; Tang, S.C.; Lee, J.E.; Lai, D.M.; Huang, S.J.; Hsieh, S.T.; Jeng, J.S.; Tu, Y.K. Prognostic value of intrathecal heme oxygenase-1 concentration in patients with Fisher Grade III aneurysmal subarachnoid hemorrhage. J. Neurosurg. 2014, 121, 1388–1393.
- Frase, S.; Steimer, M.; Selzner, L.; Kaiser, S.; Foit, N.A.; Niesen, W.D.; Schallner, N. Temporal Expression Pattern of Hemoxygenase-1 Expression and Its Association with Vasospasm and Delayed Cerebral Ischemia After Aneurysmal Subarachnoid Hemorrhage. Neurocrit. Care 2022, 36, 279–291.
- Khey, K.M.W.; Huard, A.; Mahmoud, S.H. Inflammatory Pathways Following Subarachnoid Hemorrhage. Cell Mol. Neurobiol. 2020, 40, 675–693.
- Bozza, M.T.; Jeney, V. Pro-inflammatory Actions of Heme and Other Hemoglobin-Derived DAMPs. Front. Immunol. 2020, 11, 1323.
- Figueiredo, R.T.; Fernandez, P.L.; Mourao-Sa, D.S.; Porto, B.N.; Dutra, F.F.; Alves, L.S.; Oliveira, M.F.; Oliveira, P.L.; Graca-Souza, A.V.; Bozza, M.T. Characterization of heme as activator of Toll-like receptor 4. J. Biol. Chem. 2007, 282, 20221–20229.
- Becher, B.; Spath, S.; Goverman, J. Cytokine networks in neuroinflammation. Nat. Rev. Immunol. 2017, 17, 49–59.
- Schneider, U.C.; Schiffler, J.; Hakiy, N.; Horn, P.; Vajkoczy, P. Functional analysis of Pro-inflammatory properties within the cerebrospinal fluid after subarachnoid hemorrhage in vivo and in vitro. J. Neuroinflammation 2012, 9, 28.
- Ridwan, S.; Grote, A.; Simon, M. Interleukin 6 in cerebrospinal fluid is a biomarker for delayed cerebral ischemia (DCI) related infarctions after aneurysmal subarachnoid hemorrhage. Sci. Rep. 2021, 11, 12.
- Lenski, M.; Huge, V.; Briegel, J.; Tonn, J.C.; Schichor, C.; Thon, N. Interleukin 6 in the Cerebrospinal Fluid as a Biomarker for Onset of Vasospasm and Ventriculitis After Severe Subarachnoid Hemorrhage. World Neurosurg. 2017, 99, 132–139.
- Wu, W.; Guan, Y.; Zhao, G.; Fu, X.J.; Guo, T.Z.; Liu, Y.T.; Ren, X.L.; Wang, W.; Liu, H.R.; Li, Y.Q. Elevated IL-6 and TNF-alpha Levels in Cerebrospinal Fluid of Subarachnoid Hemorrhage Patients. Mol. Neurobiol. 2016, 53, 3277–3285.
- Hanafy, K.A.; Stuart, R.M.; Khandji, A.G.; Connolly, E.S.; Badjatia, N.; Mayer, S.A.; Schindler, C. Relationship between brain interstitial fluid tumor necrosis factor-alpha and cerebral vasospasm after aneurysmal subarachnoid hemorrhage. J. Clin. Neurosci. 2010, 17, 853–856.
- Galea, J.; Ogungbenro, K.; Hulme, S.; Patel, H.; Scarth, S.; Hoadley, M.; Illingworth, K.; McMahon, C.J.; Tzerakis, N.; King, A.T.; et al. Reduction of inflammation after administration of interleukin-1 receptor antagonist following aneurysmal subarachnoid hemorrhage: Results of the Subcutaneous Interleukin-1Ra in SAH (SCIL-SAH) study. J. Neurosurg. 2018, 128, 515–523.
- Singh, N.; Hopkins, S.J.; Hulme, S.; Galea, J.P.; Hoadley, M.; Vail, A.; Hutchinson, P.J.; Grainger, S.; Rothwell, N.J.; King, A.T.; et al. The effect of intravenous interleukin-1 receptor antagonist on inflammatory mediators in cerebrospinal fluid after subarachnoid haemorrhage: A phase II randomised controlled trial. J. Neuroinflammation 2014, 11, 1.
- Sarrafzadeh, A.; Schlenk, F.; Gericke, C.; Vajkoczy, P. Relevance of cerebral interleukin-6 after aneurysmal subarachnoid hemorrhage. Neurocrit. Care 2010, 13, 339–346.
- Smith, C.J.; Hulme, S.; Vail, A.; Heal, C.; Parry-Jones, A.R.; Scarth, S.; Hopkins, K.; Hoadley, M.; Allan, S.M.; Rothwell, N.J.; et al. SCIL-STROKE (Subcutaneous Interleukin-1 Receptor Antagonist in Ischemic Stroke): A Randomized Controlled Phase 2 Trial. Stroke 2018, 49, 1210–1216.
- Ma, C.; Zhou, W.; Yan, Z.; Qu, M.; Bu, X. Toll-like receptor 4 (TLR4) is correlated with delayed cerebral ischemia (DCI) and poor prognosis in aneurysmal subarachnoid hemorrhage. J. Neurol. Sci. 2015, 359, 67–71.
- Sokol, B.; Wasik, N.; Jankowski, R.; Holysz, M.; Wieckowska, B.; Jagodzinski, P. Soluble Toll-Like Receptors 2 and 4 in Cerebrospinal Fluid of Patients with Acute Hydrocephalus following Aneurysmal Subarachnoid Haemorrhage. PLoS ONE 2016, 11, e0156171.
- Matsunaga, N.; Tsuchimori, N.; Matsumoto, T.; Ii, M. TAK-242 (resatorvid), a small-molecule inhibitor of Toll-like receptor (TLR) 4 signaling, binds selectively to TLR4 and interferes with interactions between TLR4 and its adaptor molecules. Mol. Pharmacol. 2011, 79, 34–41.
- Okada, T.; Kawakita, F.; Nishikawa, H.; Nakano, F.; Liu, L.; Suzuki, H. Selective Toll-Like Receptor 4 Antagonists Prevent Acute Blood-Brain Barrier Disruption After Subarachnoid Hemorrhage in Mice. Mol. Neurobiol. 2019, 56, 976–985.
- Okada, T.; Lei, L.; Nishikawa, H.; Nakano, F.; Nakatsuka, Y.; Suzuki, H. TAK-242, Toll-Like Receptor 4 Antagonist, Attenuates Brain Edema in Subarachnoid Hemorrhage Mice. Acta Neurochir. Suppl. 2020, 127, 77–81.
- Liu, F.Y.; Cai, J.; Wang, C.; Ruan, W.; Guan, G.P.; Pan, H.Z.; Li, J.R.; Qian, C.; Chen, J.S.; Wang, L.; et al. Fluoxetine attenuates neuroinflammation in early brain injury after subarachnoid hemorrhage: A possible role for the regulation of TLR4/MyD88/NF-kappaB signaling pathway. J. Neuroinflammation 2018, 15, 347.
- Rahimifard, M.; Maqbool, F.; Moeini-Nodeh, S.; Niaz, K.; Abdollahi, M.; Braidy, N.; Nabavi, S.M.; Nabavi, S.F. Targeting the TLR4 signaling pathway by polyphenols: A novel therapeutic strategy for neuroinflammation. Ageing Res. Rev. 2017, 36, 11–19.
- Lee, J.W.; Ahn, J.Y.; Hasegawa, S.; Cha, B.Y.; Yonezawa, T.; Nagai, K.; Seo, H.J.; Jeon, W.B.; Woo, J.T. Inhibitory effect of luteolin on osteoclast differentiation and function. Cytotechnology 2009, 61, 125–134.
- Yang, Y.; Tan, X.; Xu, J.; Wang, T.; Liang, T.; Xu, X.; Ma, C.; Xu, Z.; Wang, W.; Li, H.; et al. Luteolin alleviates neuroinflammation via downregulating the TLR4/TRAF6/NF-kappaB pathway after intracerebral hemorrhage. Biomed Pharmacother. 2020, 126, 110044.
- Park, S.J.; Song, H.Y.; Youn, H.S. Suppression of the TRIF-dependent signaling pathway of toll-like receptors by isoliquiritigenin in RAW264.7 macrophages. Mol. Cells 2009, 28, 365–368.
- Zhu, H.T.; Bian, C.; Yuan, J.C.; Chu, W.H.; Xiang, X.; Chen, F.; Wang, C.S.; Feng, H.; Lin, J.K. Curcumin attenuates acute inflammatory injury by inhibiting the TLR4/MyD88/NF-kappaB signaling pathway in experimental traumatic brain injury. J. Neuroinflammation 2014, 11, 59.
- Sun, X.G.; Ma, Q.; Jing, G.; Wang, G.Q.; Hao, X.D.; Wang, L. Increased levels of soluble triggering receptor expressed on myeloid cells-1 in cerebrospinal fluid of subarachnoid hemorrhage patients. J. Clin. Neurosci. 2017, 35, 139–143.
- Hirsch, Y.; Geraghty, J.R.; Katz, E.A.; Testai, F.D. Inflammasome Caspase-1 Activity is Elevated in Cerebrospinal Fluid After Aneurysmal Subarachnoid Hemorrhage and Predicts Functional Outcome. Neurocrit. Care 2021, 34, 889–898.
- Wu, Q.; Wang, X.L.; Yu, Q.; Pan, H.; Zhang, X.S.; Zhang, Q.R.; Wang, H.D.; Zhang, X. Inflammasome Proteins in Cerebrospinal Fluid of Patients with Subarachnoid Hemorrhage are Biomarkers of Early Brain Injury and Functional Outcome. World Neurosurg. 2016, 94, 472–479.
- Coll, R.C.; Hill, J.R.; Day, C.J.; Zamoshnikova, A.; Boucher, D.; Massey, N.L.; Chitty, J.L.; Fraser, J.A.; Jennings, M.P.; Robertson, A.A.B.; et al. MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat. Chem. Biol. 2019, 15, 556–559.
- Ismael, S.; Zhao, L.; Nasoohi, S.; Ishrat, T. Inhibition of the NLRP3-inflammasome as a potential approach for neuroprotection after stroke. Sci. Rep. 2018, 8, 5971.
- Wang, H.; Chen, H.; Jin, J.; Liu, Q.; Zhong, D.; Li, G. Inhibition of the NLRP3 inflammasome reduces brain edema and regulates athe distribution of aquaporin-4 after cerebral ischaemia-reperfusion. Life Sci. 2020, 251, 117638.

