1. The Toll-Like Receptors (TLRs) R4 Pathway
Throughout the brain, TLR4 is located on microglia (
Figure 1)
[1][35]. It recognizes its ligands together with myeloid differentiation factor 2 (MD2). The most common ligands are lipopolysaccharides (LPS), which are structures derived from the outer membrane of Gram-negative bacteria. When binding to a ligand, two TLR4–MD2 complexes, together with the ligand, are required to build a TLR4 homodimer
[2][36].
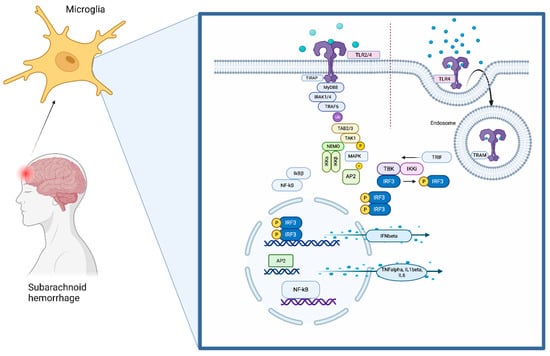
Figure 1. The TLR4 pathway. TLR4 is localized on the cell surface of microglia, where it senses damage-associated molecular patterns (DAMPs)DAMPs that are derived during brain damage. The TLR signaling pathway is activated by ligand-induced dimerization of receptors, followed by the binding of MyD88 and TIRAP or TRIF and TRAM. TLR4 translocates from the plasma membrane to endosomes with a switch from MYD88 to TRIF signaling. Activation of IRAK-1, IRAK-2, IRAK-4, and TRAFs leads to the activation of TAK1, TAK1-binding protein (TAB)1, TAB2, and TAB3. This complex phosphorylates and therefore activates IKKβ, which again forms a complex with IKKα, IKKβ, and NEMO. This leads to phosphorylation of NFκB inhibitory protein, which consequently frees NFκB. TAK1 is also involved in the phosphorylation and therefore activation of MAPKs. Transcription factors such as NFκB and AP1 induce proinflammatory cytokines. Activation of endosomal TLRs leads to the production of interferons through transcription factor IRF3. Although TLR4 has the ability to use both pathway arms, TLR2 can only signal through the MyD88-dependent pathway.
TLR4 contains N-terminal leucine-rich repeats (LRRs) and a transmembrane region, which is followed by a Toll/IL1R homology (TIR) domain. The TIR domain is connected to adaptor molecules (TIRAP). TLR4 signaling involves mainly myeloid differentiation primary response protein 88 (MyD88) and TIR-domain-containing adaptor-inducing IFN-beta (TRIF). The pathway then divides into the MyD88-dependent or the MyD88-independent pathway depending on which TIR domain is used
[3][4][5][6][6,37,38,39].
Taken together, activation of TLR4 mainly triggers inflammation through production of proinflammatory cytokines by releasing the transcription factors NFκB and AP1 into the nucleus. The mechanisms balancing the production of anti-inflammatory type-1 interferons over TRIF-mediated TLR4 signaling and the production of proinflammatory cytokines through activation of NFκB and MAPKs are not fully understood yet. Signaling protein TRAF3 might play a decisive role in determining in which direction the pathway proceeds
[7][8][40,41] Huai and colleagues reported that the tyrosine phosphatase PTPN4 dephosphorylates and, as a result, inhibits tyrosine phosphorylation of TRAM and thereby specifically inhibits the TRIF-dependent pathway
[9][42].
2. Crosstalk of TLR4 and the NLRP3 Inflammasome
When analyzing inflammatory processes associated with
subarachnoid hemorrhage (SAH
), the contribution of the inflammasome seems inevitable
[10][43]. Inflammasomes are cytosolic multiprotein complexes that can activate proinflammatory caspase-1 through autoproteolytic activation of procaspase-1. As a consequence, this leads to the cleavage of pro-inflammatory cytokines, mainly IL1β and IL18, initiating an inflammatory immune response
[11][44]. Caspase-1 also induces an inflammatory form of cell death named pyroptosis, which is highly associated with inflammasome activation
[12][45].
Currently, the best characterized inflammasome is the NLRP3 inflammasome, which consists of the NLRP3 scaffold, a sensor, an adaptor domain (ASC), and procaspase-1, the effector. ASC consists of a C-terminal caspase activation and recruitment (CARD) domain and an N-terminal pyrin domain (PYD), which enables bridging of the sensor and the effector
[11][44]. In the
central nervous system (CNS)CNS, inflammasomes are mainly present in microglia but have also been detected in neurons
[13][46]. Similarly to the TLR4 pathway, the NLRP3 inflammasome can become activated not only through pathogens but also host-derived DAMPs arising from tissue damage resulting from sterile injury as seen during SAH
[14][47].
However, the activation and signaling of inflammasomes is a complex process, where many different activators can potentially contribute.
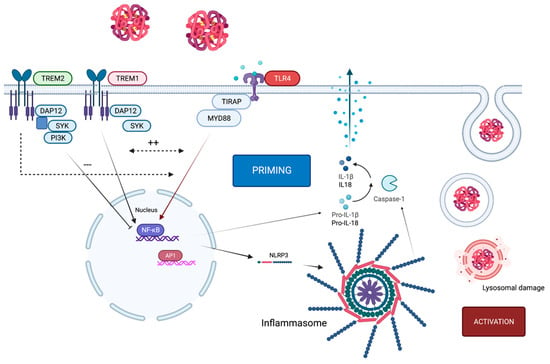
To activate an inflammasome, a two-step procedure is necessary, which requires two different signals. The priming step leads to the transduction and translation of inflammasome components (NLRP3, caspase-1, ASC, proIL1β). This first signal often appears through signaling using pattern recognition receptors (PRRs), such as TLR4, which activates NFκB to induce transcription of NLRP3 and pro-IL1β.
The second signal, the activation step, triggers the formation of the inflammasome complex. A wide range of stimuli in the cytosol have been found to initiate the activation of NLRP3, including potassium efflux, calcium accumulation, ATP, and cathepsin release through lysosomal damage or mitochondrial-injury-induced ROS production, to name but a few of the most investigated mechanisms
[13][15][46,48].
TLR4 activation through DAMPs appears to be the priming signal for inflammasome activation via activation of NFκB and therefore transcription of NLRP3 and pro-IL1β. DAMPs such as in red blood cell lysate or HMGB1 can act as signal 1 through binding to TLR4 but also as a secondary signal by binding to the inflammasome when phagocytosed by microglia
[16][17][18][49,50,51]. This leads to self-reinforcement of TLR4 and NLRP3 with exacerbation of the inflammatory response
[19][20][52,53] (
Figure 2).
Figure 2. TLR4 in context of TREM1/2 and the NLRP3 inflammasome. Activation of TLR4 through hemoglobin causes expression of the NLRP3 inflammasome, which initiates the priming step. For activation, lysosomal damage (as one example of many potential activation mechanisms) induces activation of the NLRP3 inflammasome. Upon ligand binding, the TREM2-associated adaptor DAP12 undergoes tyrosine phosphorylation by the protein kinase SRC and recruits the tyrosine protein kinase SYK, which activates phosphatidylinositol 3-kinase. TREM2 limits inflammation by interfering with proinflammatory signals, which can be transmitted by TLR4. By contrast, the TREM1 signaling cascade reinforces the production of inflammatory mediators that are initiated by TLR4 and act synergistically. TREM1 acts here as an amplifier of TLR-induced inflammation and vice versa.
Interestingly, IFNβ, also a product of the TLR4 pathway, is reported to have an immunomodulatory effect on the NLRP3 inflammasome, as it is able to suppress caspase-1 activity and therefore IL1β-expression
[21][54].
Thus, the inflammasome is dependent on TLR4. This might be a double-edged sword, as the inflammatory part of the TLR4 pathway can induce NLRP3 activation, but the immunomodulatory part, leading through IRF3 to IFNβ expression, can inhibit IL1β release.
Particularly in SAH, the NLRP3 inflammasome is reported to be part of the innate immune response appearing after the event
[22][23][24][17,55,56].
3. TLR4-Pathway-Associated Proteins Acting as Biomarkers following SAH
3.1. Ligands of TLR4
DAMPs are physiologically sequestered intracellularly and are therefore invisible to the immune system. If cells die or become damaged due to neuronal damage after SAH, these patterns then become visible to immune cells of the CNS, mainly for microglia and their receptors
[25][7]. Even though the list of potential DAMPs is growing constantly and is becoming increasingly heterogenous, their presence homogenously results in inflammation with expression of inflammation-related molecules and cytokines, iNOS, or COX2. Therefore, they are closely linked with neuroinflammatory activity after SAH and can work as biomarkers in predicting disease progression and potential anti-inflammatory therapeutic outcome
[26][88].
3.1.1. HMGB1
HMGB1 is a DNA-binding protein which is located in the nucleus of neurons. When secreted extracellularly through active release or cell death, it interacts with its main receptors TLR4 and the receptor for advanced glycation end products (RAGE) as a DAMP
[27][89].
It was shown that HMGB1 levels in the CSF and plasma of patients suffering from SAH are positively correlated not only with IL6 and TNFα levels but also with the severity and the clinical outcome
[17][28][29][30][50,90,91,92].
Reduction in mRNA and protein expression of HMGB1 upon treatment with the natural HMGB1 inhibitor glycyrrhizin was shown to improve neurological scores in an experimental model of SAH
[31][93].
HMGB1 mAB were also used in preclinical studies focusing on early brain injury after SAH, where a significant decrease in inflammatory cytokines and associated brain injury could be detected. Furthermore, delayed cerebral vasospasm was attenuated
[32][94]. This observation aligns with the findings of Nakahara and colleagues, showing that high CSF and plasma levels of HMGB1 are correlated with poor neurological outcome after SAH
[17][50]. Zhu and colleagues showed that HMGB1 is predictive of poor functional outcome after 1 year, but not for the prediction of mortality after 1 year, in-hospital mortality, or vasospasm
[30][92].
3.1.2. Red Blood Cell Lysate
During subarachnoid hemorrhage, the brain is exposed to blood particles and cell lysates of erythrocytes. Therefore, hemoglobin and its constituents moved into focus regarding initiation of an immune response by binding to TLR4 on the surface of microglia
[33][34][18,95].
Wang and colleagues measured the levels of heme oxygenase-1 (HO-1), oxyhemoglobin, ferritin, and bilirubin in intrathecal CSF on the 7th day post-hemorrhage. HO-1 appeared to be the most significant CSF parameter related to an unfavorable outcome
[35][96].
In contrast, Frase and colleagues also described increased CSF HO-1 levels in patients after SAH, but with no significant difference in functional outcome. However, lower HO-1 levels on day 7 post-SAH were associated with vasospasm
[36][97].
Oxyhemoglobin (OxyHb) is released during the lysis of red blood cells and interacts as a DAMP with TLR4
[37][34]. It autoxidizes to methemoglobin, which is also shown to be a TLR4 ligand
[16][49], as well as heme, another hemolysis-associated DAMP
[38][39][98,99]. Heme metabolism results in the breakdown products free iron and bilirubin. Free iron, in particular, is postulated to catalyze the formation of reactive oxygen species (ROS), which act as a strong inflammatory activator by binding to TLR4
[39][99].
3.2. Inflammatory Cytokines
Particularly in the CNS, where the initial immune response is triggered by microglia and in part by astrocytes, inflammatory cytokines play an emerging role when it comes to spreading inflammation in the brain
[40][100].
Interleukin (IL)-6 seems to be an important interleukin in the inflammatory response after SAH, and many studies describe a strong correlation between elevated IL6 levels in plasma and CSF and worse outcomes
[41][42][43][83,101,102].
TNFα was also described as being elevated in CSF and plasma in SAH compared with healthy controls, and higher CSF levels are linked with poorer outcomes
[44][45][103,104].
In clinical phase II studies, significantly lower levels of IL6 and CRP as inflammatory markers were observed in SAH patients after subcutaneous administration of IL1RA compared to a placebo group. However, the clinical outcome was not improved significantly
[46][47][105,106]. CSF-IL6, but not IL6, plasma levels are significantly correlated with the development of delayed brain injury after SAH, indicating that inflammation-triggered brain injury is a relevant problem not just in theory or preclinical trials but also in clinical settings
[48][107]. Analogous to the SAH studies, IL1RA was also applied subcutaneously to patients after suffering from cerebral ischemia, where diminished IL6 plasma concentrations and a better clinical outcome were observed in the treated group
[49][108].
3.3. TLR4
TLR4 itself was also investigated in SAH patients to be a potential biomarker. SAH patients were found to have higher TLR4 expression levels on the cell surface of peripheral blood mononuclear cells. TLR4 expression was determined by flow cytometry, and patients were assessed every day after admission to monitor the occurrence of delayed cerebral ischemia (DCI). Patients with DCI showed significantly higher TLR4 levels than those without DCI
[50][14]. Another study demonstrated increased levels of TLR4 in the CSF of patients 3 days after SAH
[51][109].
TAK-242 binds to the intracellular domain of TLR4 and selectively inhibits TLR4 signaling
[52][110]. It has been shown to ameliorate brain damage caused by neuroinflammation in SAH
[53][54][111,112].
Another drug which has been reported to downregulate TLR4 expression is fluoxetine. Fluoxetine is a common selective serotonin reuptake inhibitor (SSRI) that also has the ability to downregulate TLR4 and the NLRP3 inflammasome after SAH
[55][113]. However, the exact mechanisms behind the neuroprotective effects of fluoxetine remain elusive.
Polyphenols have also been described as modulators of the TLR4 pathway. They include a wide variety of molecules which all share a similar basic polyphenolic structure. They serve to protect against a variety of pathogens with anti-inflammatory and antioxidant properties
[56][114]. Resveratrol, a potential neuroprotective polyphenol, has the ability to inhibit murine RAW 264.7 macrophages und microglial BV-2 cells, and it also inhibits downstream phosphorylation of STAT1 und STAT3. Flavonoid derivates were shown to inhibit LPS-induced TLR4 dimerization in RAW 264.7 macrophage lines. Luteolin, another polyphenol, suppresses activation of IRF3 and NFκB induced by TLR4 agonists with similar effects as flavonoids or resveratrol: attenuation of inflammatory cytokines, cleaved through activation of the TRIF-dependent pathway
[57][58][59][115,116,117]. Curcumin, a polyphenolic compound, has been shown to suppress TLR4-positive microglia in the brain and therefore lead to diminished LPS-induced IRF3 activation
[60][118].
However, therapeutical approaches directly targeting TLR4 have not been performed in clinical studies but only in experimental preclinical settings.
3.4. TREM1
TREM1 is closely correlated with TLR4 pathway activation (as mentioned above). It has been investigated as one of the amplifiers of various inflammatory diseases. Concentrations of soluble TREM1 (sTREM1) have been assessed in the CSF of SAH patients. Sun and colleagues explored the correlations of early CSF sTREM1 levels with severity and prognosis and found that sTREM1 levels in CSF of SAH patients were significantly increased compared with healthy controls, and correlated negatively with Glasgow Coma Scale (GCS) and positively with the Hunt and Hess scale
[61][119]. The same group stated that inhibition of TREM1 with LP17, a synthetic TREM1 inhibitor, ameliorated microglial pyroptosis and IL1β production in experimental SAH. They also found that TREM1 can activate the NLRP3 inflammasome, which is also closely correlated with the TLR4 pathway
[14][47].
3.5. Inflammasome Proteins
The NLRP3 inflammasome plays an important role in the inflammatory process in many diseases affecting the brain
[13][46]. As its activation is closely associated with the TLR4 pathway, it should also be considered when discussing potential biomarkers and treatment strategies focusing on neuroinflammation after SAH.
Caspase-1 is a product of the inflammatory cascade found after inflammasome activation, and its occurrence in CSF was tested in 18 SAH patients. CSF analysis demonstrated a nearly seven-fold increase in caspase-1 activity in SAH patients compared with controls. Mean caspase-1 activity in the poor outcome group was approximately three times higher than the good outcome group and, furthermore, caspase-1 activity was significantly correlated with GOS score and clinical outcome
[62][120].
Another study not only assessed caspase-1 but also inflammasome proteins such as ASC and NLRP1. They also found that higher levels of inflammasome proteins were associated with severe SAH and poor outcome 3 months after SAH
[63][121].
MCC950 is a small-molecule inhibitor of the NLRP3 inflammasome that interacts with the NACHT domain and inhibits ATP hydrolysis
[64][122]. It has been found to have beneficial effects in ischemic stroke, where it leads to decreased TNFα levels, improved neurological outcome, reduced cerebral edema, and decreased infarction size
[65][66][123,124] in animal studies.