 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Shamsher Singh | -- | 3221 | 2022-11-03 10:21:07 | | | |
| 2 | Peter Tang | Meta information modification | 3221 | 2022-11-04 02:24:49 | | | | |
| 3 | Peter Tang | Meta information modification | 3221 | 2022-11-07 09:04:02 | | |
Video Upload Options
Parkinson’s disease (PD) is a neurodegenerative movement disorder characterized by the loss of dopaminergic neurons, which results in motor impairment. Ca2+ homeostasis disruption and mitochondrial dysfunction play a vital role in PD aetiology. In addition, the L-type voltage-gated calcium channel is expressed at high levels amongst nigral neurons, and could play a role in the pathogenesis of PD. In the dopaminergic neurons, Ca2+ entry through plasma membrane Cav1 channels drives a sustained feed-forward stimulation of mitochondrial oxidative phosphorylation. The R-type calcium channel is a type of voltage-dependent calcium channel. Available findings suggest that calcium homeostasis in dopaminergic neurons might be a valuable target for developing new drugs for PD patients.
1. Introduction
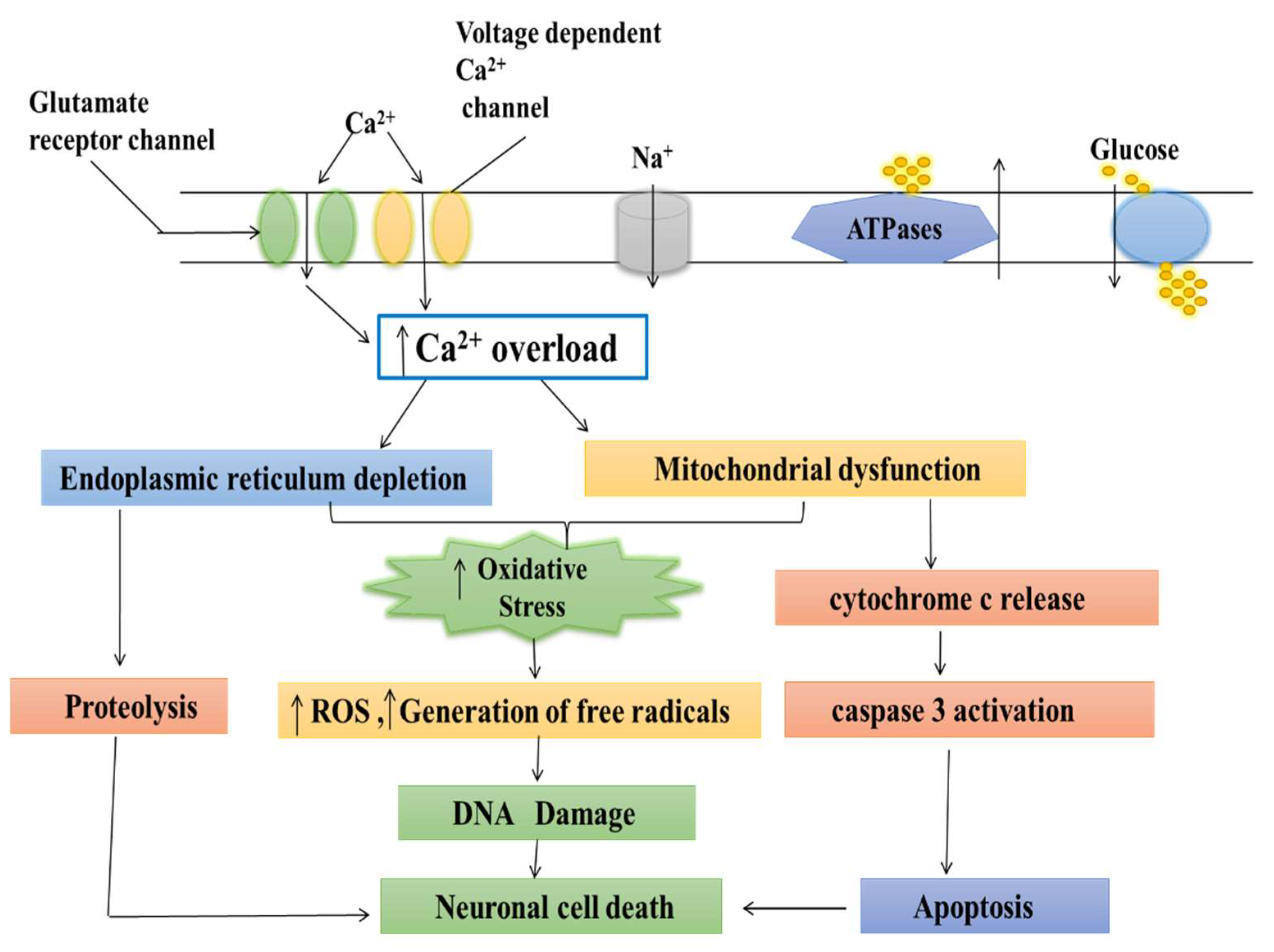
2. Normal Physiology and Pathology of Mitochondria
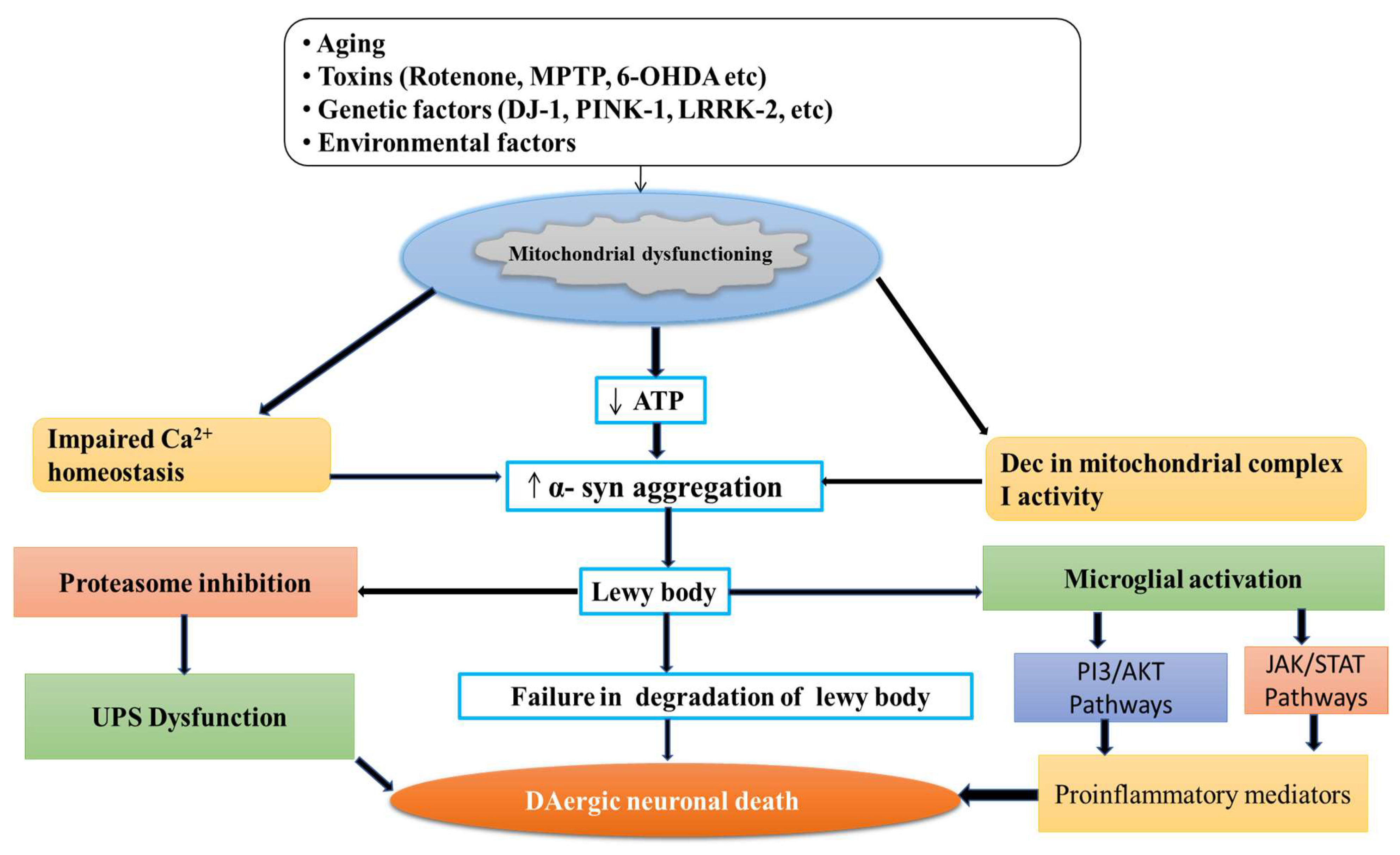
3. Role of Calcium in Mitochondria
4. Medicinal Plants as Calcium Channel Blockers
|
S. No. |
Common Name Botanical Name |
Family |
Chemical Constituents |
Plant Part Used |
|---|---|---|---|---|
|
1. |
Yarrow Achillea wilhelmsii |
Asteraceae |
Carvacrol, luteolin, apigenin 1,8-cineole |
Aerial part |
|
2. |
Shell ginger Alpinia zerumbet |
Zingiberaceae |
Catechin, epicatechin, kaempferol 3-o-rutinoside, rutin |
Whole plant |
|
3. |
Celery Apium graveolens |
Apiaceae |
Apiin, apigenin, isoquercitrin sesquiterpene |
Seed |
|
4. |
Nikko Maple Acer nikoense (Miq.) Maxim |
Aceraceae |
Scopoletin, Cleomiscosin A, Aquillochin |
Leaves, bark |
|
5. |
Soursop, Graviola Annona muricata |
Annonaceae |
Reticuline, quercetin, beta-caryophyllene, coreximine, anomurin |
Leaves |
|
6. |
Punarnava Hogweed Boerhavia diffusa |
Nyctaginaceae |
Liriodendron, boeravinone, hypoxanthine |
Whole plant, root |
|
7. |
Sweet flag, flagroot Acorus calamus L. |
Acoraceae |
β- asarone, β- gurjunene, sequesterpenes, xylose, β- daucosterol, d- galacturonic acid |
Rhizome |
|
8. |
Cape periwinkle, periwinkle Catharanthus roseus |
Apocynaceae |
Vinblastine, vincristine |
Leaves, roots, flowers |
|
9. |
Saffron Crocus sativus |
Iridaceae |
Crocin, picrocrocin, safranal, crocetin |
Stigma |
|
10. |
Carrot Daucus carota |
Apiaceae |
Coumarin glycosides (DC-2 and DC-3) |
Aerial parts |
|
11. |
Ajwain Carrom copticum |
Apiaceae |
Thymol, ρ-cymene, γ- terpinene, o-cymene, carvacrol β-phellandrene |
Seeds |
|
12. |
White horehound Marrubium vulgare L |
Lamiaceae |
Marrubenol |
Whole plant |
|
13. |
Mu Dan Pi Moutan Cortex |
Paeoniaceae |
Paeoniflorin, benzoyl paeoniflorin, mudanpioside C, paeonol, 1,2,3,4,6-o-pentagalloylglucose |
Whole plant |
|
14. |
Wu-Chu-Yu Evodia rutaecarpa L. |
Rutaceae |
Rutaecarpine |
Fruits |
|
15. |
Roselle Hibiscus sabdariffa |
Malvaceae |
β-carotene, ascorbic acid, β sitosterol, cyaniding-3- rutinose, pectin |
Calyx, leaves, corolla |
|
16. |
French Lavender Lavandula stoechas |
Lamiaceae |
Fenchone, p-cymene, lavandulyl acetate, a-pinene |
Flower and oil |
|
17. |
Olive leaf Olea africana and Olea europaea |
Oleaceae |
Oleuropein |
Leaves |
|
18. |
Ginseng Panax ginseng |
Araliaceae |
Ginsenosides Rg1, Rg3, Rh1, Re, and Rd |
Roots |
|
19. |
Basil Ocimum basilicum |
Lamiaceae |
Eugenol, α-cubebene, caryophyllene, rosmarinic, estragole |
Leaves, stem |
|
20. |
Black Cumin, Seed of Blessing |
Ranunculaceae |
Thymoquinone, dithymoquinone |
Seed |
|
21. |
Cat’s Claw herb Uncaria rhynchophylla |
Rubiaceae |
Hirsutine, rhynchophylline, isorhynchophylline |
Leaves |
|
22. |
Fen Fang Ji Radix stephaniae tetrandrae |
Menispermaceae |
Tetrandrine |
Roots |
|
23. |
Zingiber officinale |
Zingiberaceae |
Gingerol, gingerdiol, gingerdione, β-carotene, capsaicin, caffeic acid |
Rhizomes |
|
24. |
Jatamansi, Indian valerian Valeriana jatamansi |
Valerianaceae |
Jatamansika, jatamansine |
Roots, rhizomes |
References
- Jankovic, J. Parkinson’s disease: Clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry 2008, 79, 368–376.
- Nazir, M.; Al-Ansari, A.; Al-Khalifa, K.; Alhareky, M.; Gaffar, B.; Almas, K. Global prevalence of periodontal disease and lack of its surveillance. Sci. World J. 2020, 2020, 2146160.
- DeMaagd, G.; Philip, A. Parkinson’s disease and its management: Part 3: Nondopaminergic and nonpharmacological treatment options. Pharm. Ther. 2015, 40, 668.
- Cammisuli, D.M.; Cammisuli, S.M.; Fusi, J.; Franzoni, F.; Pruneti, C. Parkinson’s disease–mild cognitive impairment (PD-MCI): A useful summary of update knowledge. Front. Aging Neurosci. 2019, 11, 303.
- Narayanan, N.S.; Rodnitzky, R.L.; Uc, E.Y. Prefrontal dopamine signaling and cognitive symptoms of Parkinson’s disease. Rev. Neurosci. 2013, 24, 267–278.
- Conn, K.J.; Gao, W.; McKee, A.; Lan, M.S.; Ullman, M.D.; Eisenhauer, P.B.; Fine, R.E.; Wells, J.M. Identification of the protein disulfide isomerase family member PDIp in experimental Parkinson’s disease and Lewy body pathology. Brain Res. 2004, 1022, 164–172.
- Wakabayashi, K.; Tanji, K.; Mori, F.; Takahashi, H. The Lewy body in Parkinson’s disease: Molecules implicated in the formation and degradation of α-synuclein aggregates. Neuropathology 2007, 27, 494–506.
- Kuusisto, E.; Parkkinen, L.; Alafuzoff, I. Morphogenesis of Lewy bodies: Dissimilar incorporation of α-synuclein, ubiquitin, and p62. J. Neuropathol. Exp. Neurol. 2003, 62, 1241–1253.
- Braak, H.; Del Tredici, K. Potential pathways of abnormal tau and α-synuclein dissemination in sporadic Alzheimer’s and Parkinson’s diseases. Cold Spring Harb. Perspect. Biol. 2016, 8, a023630.
- Taipa, R.; Pereira, C.; Reis, I.; Alonso, I.; Bastos-Lima, A.; Melo-Pires, M.; Magalhães, M. DJ-1 linked parkinsonism (PARK7) is associated with Lewy body pathology. Brain 2016, 139, 1680–1687.
- MacMahon Copas, A.N.; McComish, S.F.; Fletcher, J.M.; Caldwell, M.A. The pathogenesis of Parkinson’s disease: A complex interplay between astrocytes, microglia, and T lymphocytes? Front. Neurol. 2021, 771–782.
- Wang, C.; Yang, T.; Liang, M.; Xie, J.; Song, N. Astrocyte dysfunction in Parkinson’s disease: From the perspectives of transmitted α-synuclein and genetic modulation. Transl. Neurodegener. 2021, 10, 39.
- Khasnavis, S.; Pahan, K. Cinnamon treatment upregulates neuroprotective proteins Parkin and DJ-1 and protects dopaminergic neurons in a mouse model of Parkinson’s disease. J. Neuroimmune Pharmacol. 2014, 9, 569–581.
- Heilman, P.L.; Wang, E.W.; Lewis, M.M.; Krzyzanowski, S.; Capan, C.D.; Burmeister, A.R.; Du, G.; Escobar Galvis, M.L.; Brundin, P.; Huang, X.; et al. Tryptophan metabolites are associated with symptoms and nigral pathology in parkinson’s disease. Mov. Disord. 2020, 35, 2028–2037.
- Mani, S.; Sevanan, M.; Krishnamoorthy, A.; Sekar, S. A systematic review of molecular approaches that link mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Neurol. Sci. 2021, 42, 4459–4469.
- Duarte-Jurado, A.P.; Gopar-Cuevas, Y.; Saucedo-Cardenas, O.; Loera-Arias, M.D.; Montes-de-Oca-Luna, R.; Garcia-Garcia, A.; Rodriguez-Rocha, H. Antioxidant therapeutics in Parkinson’s disease: Current challenges and opportunities. Antioxidants 2021, 10, 453.
- Harsanyiova, J.; Buday, T.; Kralova Trancikova, A. Parkinson’s disease and the gut: Future perspectives for early diagnosis. Front. Neurosci. 2020, 14, 626.
- Raj, K.; Kaur, P.; Gupta, G.D.; Singh, S. Metals associated neurodegeneration in Parkinson’s disease: Insight to physiological, pathological mechanisms and management. Neurosci. Lett. 2021, 753, 135873.
- Andrade, V.M.; Aschner, M.; Dos Santos, A.P.M. Neurotoxicity of metal mixtures. In Neurotoxicity of Metals; Springer: Berlin/Heidelberg, Germany, 2017; pp. 227–265.
- Engwa, G.A.; Ferdinand, P.U.; Nwalo, F.N.; Unachukwu, M.N. Mechanism and health effects of heavy metal toxicity in humans. Poisoning Mod. World New Tricks Old Dog 2019, 10, 70–90.
- Pinto, E.; Sigaud-kutner, T.C.; Leitao, M.A.; Okamoto, O.K.; Morse, D.; Colepicolo, P. Heavy metal–induced oxidative stress in algae 1. J. Phycol. 2003, 39, 1008–1018.
- Srivastava, S.; Singh, D.; Patel, S.; Singh, M.R. Role of enzymatic free radical scavengers in management of oxidative stress in autoimmune disorders. Int. J. Biol. Macromol. 2017, 101, 502–517.
- Jellinger, K.A. Basic mechanisms of neurodegeneration: A critical update. J. Cell. Mol. Med. 2010, 14, 457–487.
- Fu, Z.; Xi, S. The effects of heavy metals on human metabolism. Toxicol. Mech. Methods 2020, 30, 167–176.
- Sun, Q.; Li, Y.; Shi, L.; Hussain, R.; Mehmood, K.; Tang, Z.; Zhang, H. Heavy metals induced mitochondrial dysfunction in animals: Molecular mechanism of toxicity. Toxicology 2022, 21, 153136.
- Ganguly, G.; Chakrabarti, S.; Chatterjee, U.; Saso, L. Proteinopathy oxidative stress and mitochondrial dysfunction: Cross talk in Alzheimer’s disease and Parkinson’s disease. Drug Des. Dev. Ther. 2017, 11, 797.
- Langston, J.W.; Ballard, P.; Tetrud, J.W.; Irwin, I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 1983, 219, 979–980.
- Burns, R.S.; LeWitt, P.A.; Ebert, M.H.; Pakkenberg, H.; Kopin, I.J. The clinical syndrome of striatal dopamine deficiency. Parkinsonism induced by 1-methyl-4-phenyl-1,2,3,6tetrahydropyridine (MPTP). N. Engl. J. Med. 1985, 312, 1418–1421.
- Bose, A.; Beal, M.F. Mitochondrial dysfunction in Parkinson’s disease. J. Neurochem. 2016, 139, 216–231.
- Blesa, J.; Phani, S.; Jackson-Lewis, V.; Przedborski, S. Classic and new animal models of Parkinson’s disease. J. Biomed. Biotechnol. 2012, 2012, 845618.
- Jiang, P.; Dickson, D.W. Parkinson’s disease: Experimental models and reality. Acta Neuropathol. 2018, 135, 13–32.
- Rai, S.N.; Birla, H.; Singh, S.S.; Zahra, W.; Patil, R.R.; Jadhav, J.P.; Gedda, M.R.; Singh, S.P. Mucuna pruriens protects against MPTP intoxicated neuroinflammation in Parkinson’s disease through NF-κB/pAKT signaling pathways. Front. Aging Neurosci. 2017, 9, 421.
- Fubini, B.; Hubbard, A. Reactive oxygen species (ROS) and reactive nitrogen species (RNS) generation by silica in inflammation and fibrosis. Free. Radic. Biol. Med. 2003, 34, 1507–1516.
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS sources in physiological and pathological conditions. Oxidative Med. Cell. Longev. 2016, 2016, 1245049.
- Taso, O.V.; Philippou, A.; Moustogiannis, A.; Zevolis, E.; Koutsilieris, M. Lipid peroxidation products and their role in neurodegenerative diseases. Ann. Res. Hosp. 2019, 3.
- Rai, S.N.; Chaturvedi, V.K.; Singh, P.; Singh, B.K.; Singh, M.P. Mucuna pruriens in Parkinson’s and in some other diseases: Recent advancement and future prospective. 3 Biotech. 2020, 10, 522.
- Benhammou, N.; Bekkara, F.A.; Panovska, T.K. Antioxidant activity of methanolic extracts and some bioactive compounds of Atriplex halimus. Comptes Rendus Chim. 2009, 12, 1259–1266.
- Rachsee, A.; Chiranthanut, N.; Kunnaja, P.; Sireeratawong, S.; Khonsung, P.; Chansakaow, S.; Panthong, A. Mucuna pruriens (L.) DC. seed extract inhibits lipopolysaccharide-induced inflammatory responses in BV2 microglial cells. J. Ethnopharmacol. 2021, 267, 113518–113531.
- Török, N.; Tanaka, M.; Vécsei, L. Searching for peripheral biomarkers in neurodegenerative diseases: The tryptophan-kynurenine metabolic pathway. Int. J. Mol. Sci. 2020, 21, 9338.
- González-Sanmiguel, J.; Schuh, C.M.; Muñoz-Montesino, C.; Contreras-Kallens, P.; Aguayo, L.G.; Aguayo, S. Complex Interaction between resident microbiota and misfolded proteins: Role in neuroinflammation and neurodegeneration. Cells 2020, 9, 2476.
- Rai, S.N.; Zahra, W.; Singh, S.S.; Birla, H.; Keswani, C.; Dilnashin, H.; Rathore, A.S.; Singh, R.; Singh, R.K.; Singh, S.P. Anti-inflammatory activity of ursolic acid in MPTP-induced parkinsonian mouse model. Neurotox. Res. 2019, 36, 452–462.
- Ikeda, Y.; Murakami, A.; Ohigashi, H. Ursolic acid An anti-and pro-inflammatory triterpenoid. Mol. Nutr. Food Res. 2008, 52, 26–42.
- Checker, R.; Sandur, S.K.; Sharma, D.; Patwardhan, R.S.; Jayakumar, S.; Kohli, V.; Sethi, G.; Aggarwal, B.B.; Sainis, K.B. Potent anti-inflammatory activity of ursolic acid, a triterpenoid antioxidant, is mediated through suppression of NF-κB, AP-1 and NF-AT. PLoS ONE 2012, 7, 31318–31334.
- Ayelign, A.; Sabally, K. Determination of chlorogenic acids (CGA) in coffee beans using HPLC. Am. J. Res. Commun. 2013, 1, 78–91.
- Clifford, M.N. Chlorogenic acids and other cinnamates–nature, occurrence, dietary burden, absorption and metabolism. J. Sci. Food Agric. 2000, 80, 1033–1043.
- Kuhnert, N.; Karaköse, H.; Jaiswal, R. Analysis of chlorogenic acids and other hydroxycinnamates in food, plants and pharmacokinetic studies. In Handbook of Analysis of Active Compounds in Functional Foods; CRC Press: Boca Raton, FL, USA, 2012; pp. 1–52.
- Jantas, D.; Chwastek, J.; Malarz, J.; Stojakowska, A.; Lasoń, W. Neuroprotective effects of methyl caffeate against hydrogen peroxide-induced cell damage: Involvement of caspase 3 and cathepsin D inhibition. Biomolecules 2020, 10, 1530.
- Singh, S.S.; Rai, S.N.; Birla, H.; Zahra, W.; Kumar, G.; Gedda, M.R.; Tiwari, N.; Patnaik, R.; Singh, R.K.; Singh, S.P. Effect of chlorogenic acid supplementation in MPTP-intoxicated mouse. Front. Pharmacol. 2018, 9, 757.
- Singh, S.S.; Rai, S.N.; Birla, H.; Zahra, W.; Rathore, A.S.; Singh, S.P. NF-κB-mediated neuroinflammation in Parkinson’s disease and potential therapeutic effect of polyphenols. Neurotox. Res. 2020, 37, 491–507.
- Giordano, R.; Saii, Z.; Fredsgaard, M.; Hulkko, L.S.; Poulsen, T.B.; Thomsen, M.E.; Henneberg, N.; Zucolotto, S.M.; Arendt-Nielsen, L.; Papenbrock, J.; et al. Pharmacological insights into halophyte bioactive extract action on anti-inflammatory, pain relief and antibiotics-type mechanisms. Molecules 2021, 26, 3140.
- Vakifahmetoglu-Norberg, H.; Ouchida, A.T.; Norberg, E. The role of mitochondria in metabolism and cell death. Biochem. Biophys. Res. Commun. 2017, 482, 426–431.
- Van Aken, O.; Van Breusegem, F. Licensed to kill: Mitochondria, chloroplasts, and cell death. Trends Plant Sci. 2015, 20, 754–766.
- Azzam, E.I.; Jay-Gerin, J.P.; Pain, D. Ionizing radiation-induced metabolic oxidative stress and prolonged cell injury. Cancer Lett. 2012, 327, 48–60.
- Valenti, D.; de Bari, L.; De Filippis, B.; Henrion-Caude, A.; Vacca, R.A. Mitochondrial dysfunction as a central actor in intellectual disability-related diseases: An overview of Down syndrome, autism, Fragile X and Rett syndrome. Neurosci. Biobehav. Rev. 2014, 46, 202–217.
- Lesage, S.; Brice, A. Role of Mendelian genes in “sporadic” Parkinson’s disease. Park. Relat. Disord. 2012, 18, S66–S70.
- Hernandez, D.G.; Reed, X.; Singleton, A.B. Genetics in Parkinson disease: Mendelian versus non-Mendelian inheritance. J. Neurochem. 2016, 139, 59–74.
- Panov, A.; Dikalov, S.; Shalbuyeva, N.; Taylor, G.; Sherer, T.; Greenamyre, J.T. Rotenone model of Parkinson disease: Multiple brain mitochondria dysfunctions after short term systemic rotenone intoxication. J. Biol. Chem. 2005, 280, 42026–42035.
- Borland, M.K.; Trimmer, P.A.; Rubinstein, J.D.; Keeney, P.M.; Mohanakumar, K.; Liu, L.; Bennett, J.P. Chronic, lowdose rotenone reproduces Lewy neurites found in early stages of Parkinson’s disease, reduces mitochondrial movement and slowly kills differentiated SH-SY5Y neural cells. Mol. Neurodegener. 2008, 3, 21.
- Franco, R.; Li, S.; Rodriguez-Rocha, H.; Burns, M.; Panayiotidis, M.I. Molecular mechanisms of pesticide-induced neurotoxicity: Relevance to Parkinson’s disease. Chem. Biol. Interact. 2010, 188, 289–300.
- Prakash, J.; Yadav, S.K.; Chouhan, S.; Singh, S.P. Neuroprotective role of Withania somnifera root extract in Maneb–Paraquat induced mouse model of parkinsonism. Neurochem. Res. 2013, 38, 972–980.
- Dinis-Oliveira, R.J.; Remiao, F.; Carmo, H.; Duarte, J.A.; Navarro, A.S.; Bastos, M.L.; Carvalho, F. Paraquat exposure as an etiological factor of Parkinson’s disease. Neurotoxicology 2006, 27, 1110–1122.
- Colle, D.; Farina, M. Oxidative stress in paraquat-induced damage to nervous tissues. In Toxicology; Academic Press: Cambridge, MA, USA, 2021; pp. 69–78.
- Gupta, S.P.; Patel, S.; Yadav, S.; Singh, A.K.; Singh, S.; Singh, M.P. Involvement of nitric oxide in maneb-and paraquat-induced Parkinson’s disease phenotype in mouse: Is there any link with lipid peroxidation? Neurochem. Res. 2010, 35, 1206–1213.
- Ahmad, I.; Kumar, A.; Shukla, S.; Prasad Pandey, H.; Singh, C. The involvement of nitric oxide in maneb-and paraquat-induced oxidative stress in rat polymorphonuclear leukocytes. Free. Radic. Res. 2008, 42, 849–862.
- Sharma, V.; Sharma, S.; Pracheta, R.P. Withania somnifera: A rejuvenating ayurvedic medicinal herb for the treatment. Int. J. Pharm. Tech. Res. 2011, 3, 187–192.
- Singh, N.; Bhalla, M.; de Jager, P.; Gilca, M. An overview on ashwagandha: A Rasayana (rejuvenator) of Ayurveda. Afr. J. Tradit. Complement. Altern. Med. 2011, 8, 208–213.
- Vegh, C.; Wear, D.; Okaj, I.; Huggard, R.; Culmone, L.; Eren, S.; Cohen, J.; Rishi, A.K.; Pandey, S. Combined Ubisol-Q10 and Ashwagandha Root Extract Target Multiple Biochemical Mechanisms and Reduces Neurodegeneration in a Paraquat-Induced Rat Model of Parkinson’s Disease. Antioxidants 2021, 10, 563.
- Gleichmann, M.; Mattson, M.P. Neuronal calcium homeostasis and dysregulation. Antioxid. Redox Signal 2011, 14, 1261–1273.
- Schapira, A.H. Mitochondria in the aetiology and pathogenesis of Parkinson’s disease. Lancet Neurol. 2008, 7, 97–109.
- Martinez, T.N.; Greenamyre, J.T. Toxin models of mitochondrial dysfunction in Parkinson’s disease. Antioxid. Redox Signal. 2012, 16, 920–934.
- Kristián, T. Metabolic stages, mitochondria and calcium in hypoxic/ischemic brain damage. Cell Calcium 2004, 36, 221–233.
- Duchen, M.R. Contributions of mitochondria to animal physiology: From homeostatic sensor to calcium signalling and cell death. J. Physiol. 1999, 516, 1–7.
- Halestrap, A.P.; Pasdois, P. The role of the mitochondrial permeability transition pore in heart disease. Biochim. Et Biophys. Acta Bioenerg. 2009, 1787, 1402–1415.
- Duchen, M.R. Roles of mitochondria in health and disease. Diabetes 2004, 53, S96–S102.
- Tang, L.; Gamal El-Din, T.M.; Payandeh, J.; Martinez, G.Q.; Heard, T.M.; Scheuer, T.; Zheng, N.; Catterall, W.A. Structural basis for Ca2+ selectivity of a voltage-gated calcium channel. Nature 2014, 505, 56–61.
- Vafai, S.B.; Mootha, V.K. Mitochondrial disorders as windows into an ancient organelle. Nature 2012, 491, 374–383.
- Pivovarova, N.B.; Andrews, S.B. Calcium-dependent mitochondrial function and dysfunction in neurons. FEBS J. 2010, 277, 3622–3636.
- Moon, H.E.; Paek, S.H. Mitochondrial dysfunction in Parkinson’s disease. Exp. Neurobiol. 2015, 24, 103–118.
- Cullen, P.J.; Lockyer, P.J. Integration of calcium and Ras signalling. Nat. Rev. Mol. Cell Biol. 2002, 3, 339–348.
- Clapham, D.E. Calcium signaling. Cell 2007, 131, 1047–1058.
- Verma, M.; Lizama, B.N.; Chu, C.T. Excitotoxicity, calcium and mitochondria: A triad in synaptic neurodegeneration. Transl. Neurodegener. 2022, 11, 3.
- Khatri, N.; Thakur, M.; Pareek, V.; Kumar, S.; Sharma, S.; Datusalia, A.K. Oxidative stress: Major threat in traumatic brain injury. CNS Neurol. Disord. Drug Targets 2018, 17, 689–695.
- Marjina Singh, A.; Sharma, A.; Narang, R.K.; Singh, G. Management of Hypertension with Conventional and Herbals Drugs. J. Drug Deliv. Ther. 2020, 10, 280–287.
- Joshi, N.J.; Shelke, S.A. Medicinal Plants as Calcium-channel Blockers Against Hypertension. Vascular 2021, 1, 4–5.



