Parkinson’s disease (PD) is a neurodegenerative movement disorder characterized by the loss of dopaminergic neurons, which results in motor impairment. Ca2+ homeostasis disruption and mitochondrial dysfunction play a vital role in PD aetiology. In addition, the L-type voltage-gated calcium channel is expressed at high levels amongst nigral neurons, and could play a role in the pathogenesis of PD. In the dopaminergic neurons, Ca2+ entry through plasma membrane Cav1 channels drives a sustained feed-forward stimulation of mitochondrial oxidative phosphorylation. The R-type calcium channel is a type of voltage-dependent calcium channel. Available findings suggest that calcium homeostasis in dopaminergic neurons might be a valuable target for developing new drugs for PD patients.
- Parkinson’s disease
- dopaminergic neurons
- mitochondrial
- calcium channel
- calcium channel blocker
- neurodegenerative disorder
- mitochondrial dysfunction
- oxidative stress
- neuroinflammation
- Lewy bodies
- dihydropyridine
1. Introduction
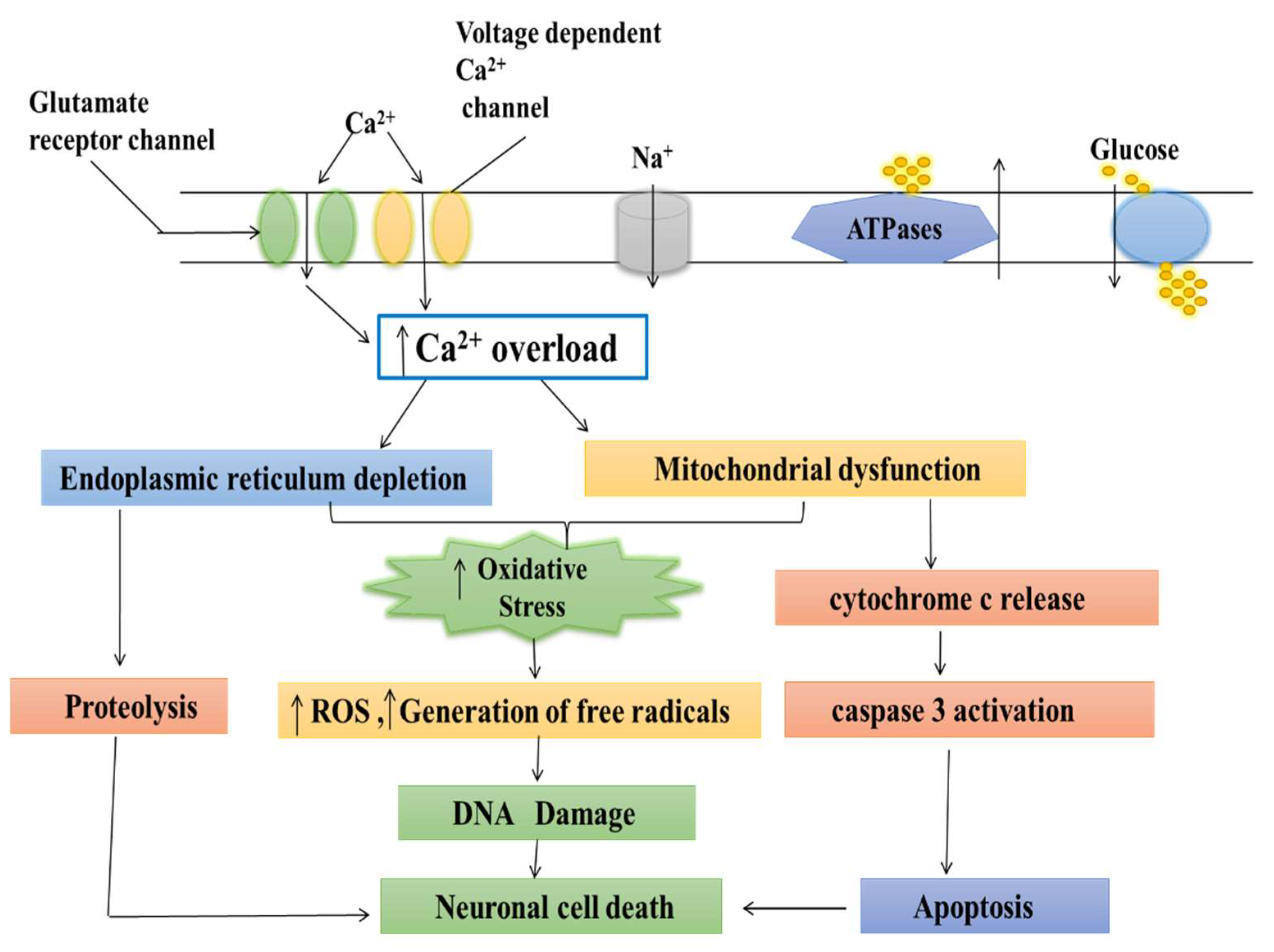
2. Normal Physiology and Pathology of Mitochondria
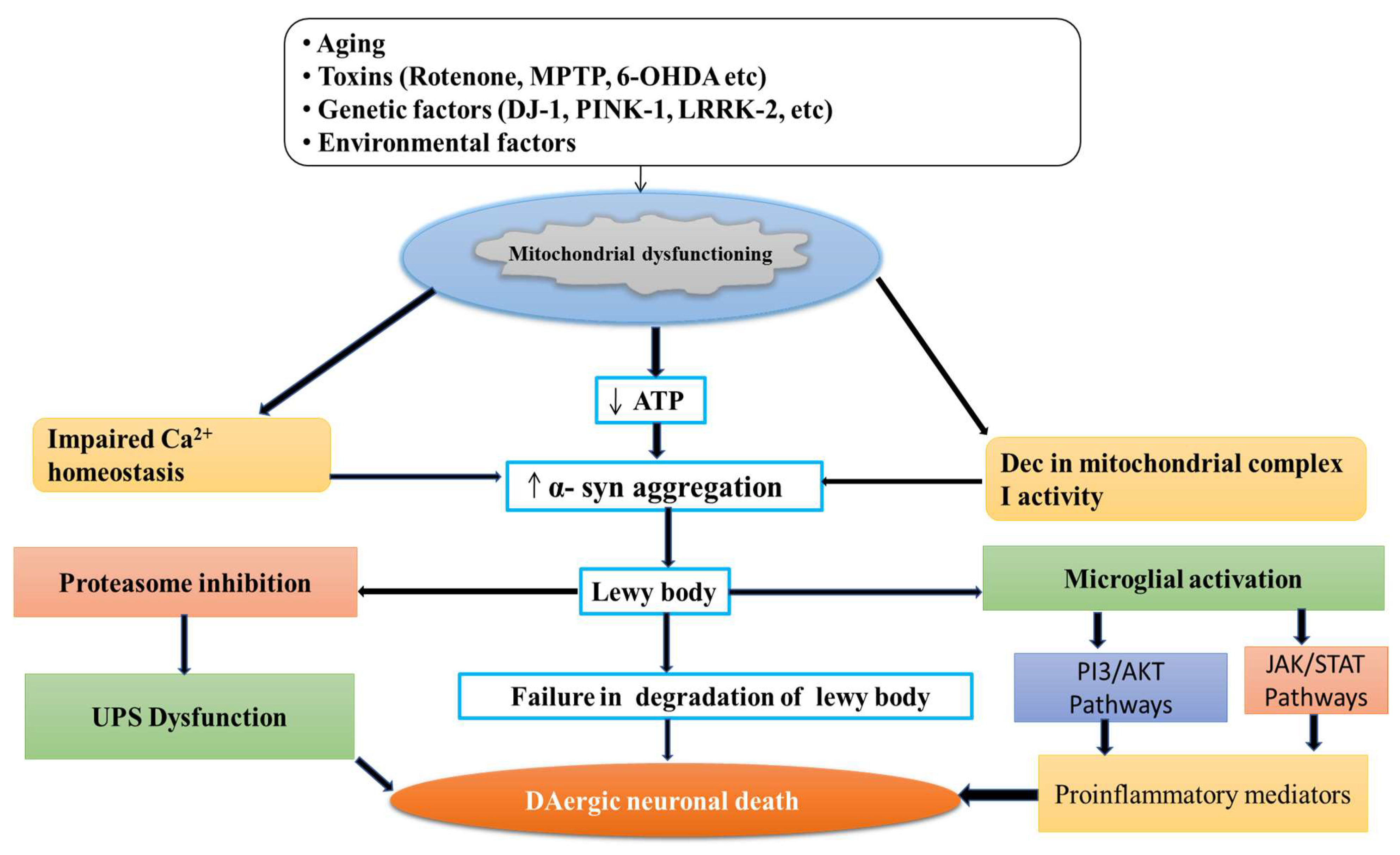
3. Role of Calcium in Mitochondria
4. Medicinal Plants as Calcium Channel Blockers
|
S. No. |
Common Name Botanical Name |
Family |
Chemical Constituents |
Plant Part Used |
|---|---|---|---|---|
|
1. |
Yarrow Achillea wilhelmsii |
Asteraceae |
Carvacrol, luteolin, apigenin 1,8-cineole |
Aerial part |
|
2. |
Shell ginger Alpinia zerumbet |
Zingiberaceae |
Catechin, epicatechin, kaempferol 3-o-rutinoside, rutin |
Whole plant |
|
3. |
Celery Apium graveolens |
Apiaceae |
Apiin, apigenin, isoquercitrin sesquiterpene |
Seed |
|
4. |
Nikko Maple Acer nikoense (Miq.) Maxim |
Aceraceae |
Scopoletin, Cleomiscosin A, Aquillochin |
Leaves, bark |
|
5. |
Soursop, Graviola Annona muricata |
Annonaceae |
Reticuline, quercetin, beta-caryophyllene, coreximine, anomurin |
Leaves |
|
6. |
Punarnava Hogweed Boerhavia diffusa |
Nyctaginaceae |
Liriodendron, boeravinone, hypoxanthine |
Whole plant, root |
|
7. |
Sweet flag, flagroot Acorus calamus L. |
Acoraceae |
β- asarone, β- gurjunene, sequesterpenes, xylose, β- daucosterol, d- galacturonic acid |
Rhizome |
|
8. |
Cape periwinkle, periwinkle Catharanthus roseus |
Apocynaceae |
Vinblastine, vincristine |
Leaves, roots, flowers |
|
9. |
Saffron Crocus sativus |
Iridaceae |
Crocin, picrocrocin, safranal, crocetin |
Stigma |
|
10. |
Carrot Daucus carota |
Apiaceae |
Coumarin glycosides (DC-2 and DC-3) |
Aerial parts |
|
11. |
Ajwain Carrom copticum |
Apiaceae |
Thymol, ρ-cymene, γ- terpinene, o-cymene, carvacrol β-phellandrene |
Seeds |
|
12. |
White horehound Marrubium vulgare L |
Lamiaceae |
Marrubenol |
Whole plant |
|
13. |
Mu Dan Pi Moutan Cortex |
Paeoniaceae |
Paeoniflorin, benzoyl paeoniflorin, mudanpioside C, paeonol, 1,2,3,4,6-o-pentagalloylglucose |
Whole plant |
|
14. |
Wu-Chu-Yu Evodia rutaecarpa L. |
Rutaceae |
Rutaecarpine |
Fruits |
|
15. |
Roselle Hibiscus sabdariffa |
Malvaceae |
β-carotene, ascorbic acid, β sitosterol, cyaniding-3- rutinose, pectin |
Calyx, leaves, corolla |
|
16. |
French Lavender Lavandula stoechas |
Lamiaceae |
Fenchone, p-cymene, lavandulyl acetate, a-pinene |
Flower and oil |
|
17. |
Olive leaf Olea africana and Olea europaea |
Oleaceae |
Oleuropein |
Leaves |
|
18. |
Ginseng Panax ginseng |
Araliaceae |
Ginsenosides Rg1, Rg3, Rh1, Re, and Rd |
Roots |
|
19. |
Basil Ocimum basilicum |
Lamiaceae |
Eugenol, α-cubebene, caryophyllene, rosmarinic, estragole |
Leaves, stem |
|
20. |
Black Cumin, Seed of Blessing |
Ranunculaceae |
Thymoquinone, dithymoquinone |
Seed |
|
21. |
Cat’s Claw herb Uncaria rhynchophylla |
Rubiaceae |
Hirsutine, rhynchophylline, isorhynchophylline |
Leaves |
|
22. |
Fen Fang Ji Radix stephaniae tetrandrae |
Menispermaceae |
Tetrandrine |
Roots |
|
23. |
Zingiber officinale |
Zingiberaceae |
Gingerol, gingerdiol, gingerdione, β-carotene, capsaicin, caffeic acid |
Rhizomes |
|
24. |
Jatamansi, Indian valerian Valeriana jatamansi |
Valerianaceae |
Jatamansika, jatamansine |
Roots, rhizomes |
This entry is adapted from the peer-reviewed paper 10.3390/jmp3040021
References
- Jankovic, J. Parkinson’s disease: Clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry 2008, 79, 368–376.
- Nazir, M.; Al-Ansari, A.; Al-Khalifa, K.; Alhareky, M.; Gaffar, B.; Almas, K. Global prevalence of periodontal disease and lack of its surveillance. Sci. World J. 2020, 2020, 2146160.
- DeMaagd, G.; Philip, A. Parkinson’s disease and its management: Part 3: Nondopaminergic and nonpharmacological treatment options. Pharm. Ther. 2015, 40, 668.
- Cammisuli, D.M.; Cammisuli, S.M.; Fusi, J.; Franzoni, F.; Pruneti, C. Parkinson’s disease–mild cognitive impairment (PD-MCI): A useful summary of update knowledge. Front. Aging Neurosci. 2019, 11, 303.
- Narayanan, N.S.; Rodnitzky, R.L.; Uc, E.Y. Prefrontal dopamine signaling and cognitive symptoms of Parkinson’s disease. Rev. Neurosci. 2013, 24, 267–278.
- Conn, K.J.; Gao, W.; McKee, A.; Lan, M.S.; Ullman, M.D.; Eisenhauer, P.B.; Fine, R.E.; Wells, J.M. Identification of the protein disulfide isomerase family member PDIp in experimental Parkinson’s disease and Lewy body pathology. Brain Res. 2004, 1022, 164–172.
- Wakabayashi, K.; Tanji, K.; Mori, F.; Takahashi, H. The Lewy body in Parkinson’s disease: Molecules implicated in the formation and degradation of α-synuclein aggregates. Neuropathology 2007, 27, 494–506.
- Kuusisto, E.; Parkkinen, L.; Alafuzoff, I. Morphogenesis of Lewy bodies: Dissimilar incorporation of α-synuclein, ubiquitin, and p62. J. Neuropathol. Exp. Neurol. 2003, 62, 1241–1253.
- Braak, H.; Del Tredici, K. Potential pathways of abnormal tau and α-synuclein dissemination in sporadic Alzheimer’s and Parkinson’s diseases. Cold Spring Harb. Perspect. Biol. 2016, 8, a023630.
- Taipa, R.; Pereira, C.; Reis, I.; Alonso, I.; Bastos-Lima, A.; Melo-Pires, M.; Magalhães, M. DJ-1 linked parkinsonism (PARK7) is associated with Lewy body pathology. Brain 2016, 139, 1680–1687.
- MacMahon Copas, A.N.; McComish, S.F.; Fletcher, J.M.; Caldwell, M.A. The pathogenesis of Parkinson’s disease: A complex interplay between astrocytes, microglia, and T lymphocytes? Front. Neurol. 2021, 771–782.
- Wang, C.; Yang, T.; Liang, M.; Xie, J.; Song, N. Astrocyte dysfunction in Parkinson’s disease: From the perspectives of transmitted α-synuclein and genetic modulation. Transl. Neurodegener. 2021, 10, 39.
- Khasnavis, S.; Pahan, K. Cinnamon treatment upregulates neuroprotective proteins Parkin and DJ-1 and protects dopaminergic neurons in a mouse model of Parkinson’s disease. J. Neuroimmune Pharmacol. 2014, 9, 569–581.
- Heilman, P.L.; Wang, E.W.; Lewis, M.M.; Krzyzanowski, S.; Capan, C.D.; Burmeister, A.R.; Du, G.; Escobar Galvis, M.L.; Brundin, P.; Huang, X.; et al. Tryptophan metabolites are associated with symptoms and nigral pathology in parkinson’s disease. Mov. Disord. 2020, 35, 2028–2037.
- Mani, S.; Sevanan, M.; Krishnamoorthy, A.; Sekar, S. A systematic review of molecular approaches that link mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Neurol. Sci. 2021, 42, 4459–4469.
- Duarte-Jurado, A.P.; Gopar-Cuevas, Y.; Saucedo-Cardenas, O.; Loera-Arias, M.D.; Montes-de-Oca-Luna, R.; Garcia-Garcia, A.; Rodriguez-Rocha, H. Antioxidant therapeutics in Parkinson’s disease: Current challenges and opportunities. Antioxidants 2021, 10, 453.
- Harsanyiova, J.; Buday, T.; Kralova Trancikova, A. Parkinson’s disease and the gut: Future perspectives for early diagnosis. Front. Neurosci. 2020, 14, 626.
- Raj, K.; Kaur, P.; Gupta, G.D.; Singh, S. Metals associated neurodegeneration in Parkinson’s disease: Insight to physiological, pathological mechanisms and management. Neurosci. Lett. 2021, 753, 135873.
- Andrade, V.M.; Aschner, M.; Dos Santos, A.P.M. Neurotoxicity of metal mixtures. In Neurotoxicity of Metals; Springer: Berlin/Heidelberg, Germany, 2017; pp. 227–265.
- Engwa, G.A.; Ferdinand, P.U.; Nwalo, F.N.; Unachukwu, M.N. Mechanism and health effects of heavy metal toxicity in humans. Poisoning Mod. World New Tricks Old Dog 2019, 10, 70–90.
- Pinto, E.; Sigaud-kutner, T.C.; Leitao, M.A.; Okamoto, O.K.; Morse, D.; Colepicolo, P. Heavy metal–induced oxidative stress in algae 1. J. Phycol. 2003, 39, 1008–1018.
- Srivastava, S.; Singh, D.; Patel, S.; Singh, M.R. Role of enzymatic free radical scavengers in management of oxidative stress in autoimmune disorders. Int. J. Biol. Macromol. 2017, 101, 502–517.
- Jellinger, K.A. Basic mechanisms of neurodegeneration: A critical update. J. Cell. Mol. Med. 2010, 14, 457–487.
- Fu, Z.; Xi, S. The effects of heavy metals on human metabolism. Toxicol. Mech. Methods 2020, 30, 167–176.
- Sun, Q.; Li, Y.; Shi, L.; Hussain, R.; Mehmood, K.; Tang, Z.; Zhang, H. Heavy metals induced mitochondrial dysfunction in animals: Molecular mechanism of toxicity. Toxicology 2022, 21, 153136.
- Ganguly, G.; Chakrabarti, S.; Chatterjee, U.; Saso, L. Proteinopathy oxidative stress and mitochondrial dysfunction: Cross talk in Alzheimer’s disease and Parkinson’s disease. Drug Des. Dev. Ther. 2017, 11, 797.
- Langston, J.W.; Ballard, P.; Tetrud, J.W.; Irwin, I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 1983, 219, 979–980.
- Burns, R.S.; LeWitt, P.A.; Ebert, M.H.; Pakkenberg, H.; Kopin, I.J. The clinical syndrome of striatal dopamine deficiency. Parkinsonism induced by 1-methyl-4-phenyl-1,2,3,6tetrahydropyridine (MPTP). N. Engl. J. Med. 1985, 312, 1418–1421.
- Bose, A.; Beal, M.F. Mitochondrial dysfunction in Parkinson’s disease. J. Neurochem. 2016, 139, 216–231.
- Blesa, J.; Phani, S.; Jackson-Lewis, V.; Przedborski, S. Classic and new animal models of Parkinson’s disease. J. Biomed. Biotechnol. 2012, 2012, 845618.
- Jiang, P.; Dickson, D.W. Parkinson’s disease: Experimental models and reality. Acta Neuropathol. 2018, 135, 13–32.
- Rai, S.N.; Birla, H.; Singh, S.S.; Zahra, W.; Patil, R.R.; Jadhav, J.P.; Gedda, M.R.; Singh, S.P. Mucuna pruriens protects against MPTP intoxicated neuroinflammation in Parkinson’s disease through NF-κB/pAKT signaling pathways. Front. Aging Neurosci. 2017, 9, 421.
- Fubini, B.; Hubbard, A. Reactive oxygen species (ROS) and reactive nitrogen species (RNS) generation by silica in inflammation and fibrosis. Free. Radic. Biol. Med. 2003, 34, 1507–1516.
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS sources in physiological and pathological conditions. Oxidative Med. Cell. Longev. 2016, 2016, 1245049.
- Taso, O.V.; Philippou, A.; Moustogiannis, A.; Zevolis, E.; Koutsilieris, M. Lipid peroxidation products and their role in neurodegenerative diseases. Ann. Res. Hosp. 2019, 3.
- Rai, S.N.; Chaturvedi, V.K.; Singh, P.; Singh, B.K.; Singh, M.P. Mucuna pruriens in Parkinson’s and in some other diseases: Recent advancement and future prospective. 3 Biotech. 2020, 10, 522.
- Benhammou, N.; Bekkara, F.A.; Panovska, T.K. Antioxidant activity of methanolic extracts and some bioactive compounds of Atriplex halimus. Comptes Rendus Chim. 2009, 12, 1259–1266.
- Rachsee, A.; Chiranthanut, N.; Kunnaja, P.; Sireeratawong, S.; Khonsung, P.; Chansakaow, S.; Panthong, A. Mucuna pruriens (L.) DC. seed extract inhibits lipopolysaccharide-induced inflammatory responses in BV2 microglial cells. J. Ethnopharmacol. 2021, 267, 113518–113531.
- Török, N.; Tanaka, M.; Vécsei, L. Searching for peripheral biomarkers in neurodegenerative diseases: The tryptophan-kynurenine metabolic pathway. Int. J. Mol. Sci. 2020, 21, 9338.
- González-Sanmiguel, J.; Schuh, C.M.; Muñoz-Montesino, C.; Contreras-Kallens, P.; Aguayo, L.G.; Aguayo, S. Complex Interaction between resident microbiota and misfolded proteins: Role in neuroinflammation and neurodegeneration. Cells 2020, 9, 2476.
- Rai, S.N.; Zahra, W.; Singh, S.S.; Birla, H.; Keswani, C.; Dilnashin, H.; Rathore, A.S.; Singh, R.; Singh, R.K.; Singh, S.P. Anti-inflammatory activity of ursolic acid in MPTP-induced parkinsonian mouse model. Neurotox. Res. 2019, 36, 452–462.
- Ikeda, Y.; Murakami, A.; Ohigashi, H. Ursolic acid An anti-and pro-inflammatory triterpenoid. Mol. Nutr. Food Res. 2008, 52, 26–42.
- Checker, R.; Sandur, S.K.; Sharma, D.; Patwardhan, R.S.; Jayakumar, S.; Kohli, V.; Sethi, G.; Aggarwal, B.B.; Sainis, K.B. Potent anti-inflammatory activity of ursolic acid, a triterpenoid antioxidant, is mediated through suppression of NF-κB, AP-1 and NF-AT. PLoS ONE 2012, 7, 31318–31334.
- Ayelign, A.; Sabally, K. Determination of chlorogenic acids (CGA) in coffee beans using HPLC. Am. J. Res. Commun. 2013, 1, 78–91.
- Clifford, M.N. Chlorogenic acids and other cinnamates–nature, occurrence, dietary burden, absorption and metabolism. J. Sci. Food Agric. 2000, 80, 1033–1043.
- Kuhnert, N.; Karaköse, H.; Jaiswal, R. Analysis of chlorogenic acids and other hydroxycinnamates in food, plants and pharmacokinetic studies. In Handbook of Analysis of Active Compounds in Functional Foods; CRC Press: Boca Raton, FL, USA, 2012; pp. 1–52.
- Jantas, D.; Chwastek, J.; Malarz, J.; Stojakowska, A.; Lasoń, W. Neuroprotective effects of methyl caffeate against hydrogen peroxide-induced cell damage: Involvement of caspase 3 and cathepsin D inhibition. Biomolecules 2020, 10, 1530.
- Singh, S.S.; Rai, S.N.; Birla, H.; Zahra, W.; Kumar, G.; Gedda, M.R.; Tiwari, N.; Patnaik, R.; Singh, R.K.; Singh, S.P. Effect of chlorogenic acid supplementation in MPTP-intoxicated mouse. Front. Pharmacol. 2018, 9, 757.
- Singh, S.S.; Rai, S.N.; Birla, H.; Zahra, W.; Rathore, A.S.; Singh, S.P. NF-κB-mediated neuroinflammation in Parkinson’s disease and potential therapeutic effect of polyphenols. Neurotox. Res. 2020, 37, 491–507.
- Giordano, R.; Saii, Z.; Fredsgaard, M.; Hulkko, L.S.; Poulsen, T.B.; Thomsen, M.E.; Henneberg, N.; Zucolotto, S.M.; Arendt-Nielsen, L.; Papenbrock, J.; et al. Pharmacological insights into halophyte bioactive extract action on anti-inflammatory, pain relief and antibiotics-type mechanisms. Molecules 2021, 26, 3140.
- Vakifahmetoglu-Norberg, H.; Ouchida, A.T.; Norberg, E. The role of mitochondria in metabolism and cell death. Biochem. Biophys. Res. Commun. 2017, 482, 426–431.
- Van Aken, O.; Van Breusegem, F. Licensed to kill: Mitochondria, chloroplasts, and cell death. Trends Plant Sci. 2015, 20, 754–766.
- Azzam, E.I.; Jay-Gerin, J.P.; Pain, D. Ionizing radiation-induced metabolic oxidative stress and prolonged cell injury. Cancer Lett. 2012, 327, 48–60.
- Valenti, D.; de Bari, L.; De Filippis, B.; Henrion-Caude, A.; Vacca, R.A. Mitochondrial dysfunction as a central actor in intellectual disability-related diseases: An overview of Down syndrome, autism, Fragile X and Rett syndrome. Neurosci. Biobehav. Rev. 2014, 46, 202–217.
- Lesage, S.; Brice, A. Role of Mendelian genes in “sporadic” Parkinson’s disease. Park. Relat. Disord. 2012, 18, S66–S70.
- Hernandez, D.G.; Reed, X.; Singleton, A.B. Genetics in Parkinson disease: Mendelian versus non-Mendelian inheritance. J. Neurochem. 2016, 139, 59–74.
- Panov, A.; Dikalov, S.; Shalbuyeva, N.; Taylor, G.; Sherer, T.; Greenamyre, J.T. Rotenone model of Parkinson disease: Multiple brain mitochondria dysfunctions after short term systemic rotenone intoxication. J. Biol. Chem. 2005, 280, 42026–42035.
- Borland, M.K.; Trimmer, P.A.; Rubinstein, J.D.; Keeney, P.M.; Mohanakumar, K.; Liu, L.; Bennett, J.P. Chronic, lowdose rotenone reproduces Lewy neurites found in early stages of Parkinson’s disease, reduces mitochondrial movement and slowly kills differentiated SH-SY5Y neural cells. Mol. Neurodegener. 2008, 3, 21.
- Franco, R.; Li, S.; Rodriguez-Rocha, H.; Burns, M.; Panayiotidis, M.I. Molecular mechanisms of pesticide-induced neurotoxicity: Relevance to Parkinson’s disease. Chem. Biol. Interact. 2010, 188, 289–300.
- Prakash, J.; Yadav, S.K.; Chouhan, S.; Singh, S.P. Neuroprotective role of Withania somnifera root extract in Maneb–Paraquat induced mouse model of parkinsonism. Neurochem. Res. 2013, 38, 972–980.
- Dinis-Oliveira, R.J.; Remiao, F.; Carmo, H.; Duarte, J.A.; Navarro, A.S.; Bastos, M.L.; Carvalho, F. Paraquat exposure as an etiological factor of Parkinson’s disease. Neurotoxicology 2006, 27, 1110–1122.
- Colle, D.; Farina, M. Oxidative stress in paraquat-induced damage to nervous tissues. In Toxicology; Academic Press: Cambridge, MA, USA, 2021; pp. 69–78.
- Gupta, S.P.; Patel, S.; Yadav, S.; Singh, A.K.; Singh, S.; Singh, M.P. Involvement of nitric oxide in maneb-and paraquat-induced Parkinson’s disease phenotype in mouse: Is there any link with lipid peroxidation? Neurochem. Res. 2010, 35, 1206–1213.
- Ahmad, I.; Kumar, A.; Shukla, S.; Prasad Pandey, H.; Singh, C. The involvement of nitric oxide in maneb-and paraquat-induced oxidative stress in rat polymorphonuclear leukocytes. Free. Radic. Res. 2008, 42, 849–862.
- Sharma, V.; Sharma, S.; Pracheta, R.P. Withania somnifera: A rejuvenating ayurvedic medicinal herb for the treatment. Int. J. Pharm. Tech. Res. 2011, 3, 187–192.
- Singh, N.; Bhalla, M.; de Jager, P.; Gilca, M. An overview on ashwagandha: A Rasayana (rejuvenator) of Ayurveda. Afr. J. Tradit. Complement. Altern. Med. 2011, 8, 208–213.
- Vegh, C.; Wear, D.; Okaj, I.; Huggard, R.; Culmone, L.; Eren, S.; Cohen, J.; Rishi, A.K.; Pandey, S. Combined Ubisol-Q10 and Ashwagandha Root Extract Target Multiple Biochemical Mechanisms and Reduces Neurodegeneration in a Paraquat-Induced Rat Model of Parkinson’s Disease. Antioxidants 2021, 10, 563.
- Gleichmann, M.; Mattson, M.P. Neuronal calcium homeostasis and dysregulation. Antioxid. Redox Signal 2011, 14, 1261–1273.
- Schapira, A.H. Mitochondria in the aetiology and pathogenesis of Parkinson’s disease. Lancet Neurol. 2008, 7, 97–109.
- Martinez, T.N.; Greenamyre, J.T. Toxin models of mitochondrial dysfunction in Parkinson’s disease. Antioxid. Redox Signal. 2012, 16, 920–934.
- Kristián, T. Metabolic stages, mitochondria and calcium in hypoxic/ischemic brain damage. Cell Calcium 2004, 36, 221–233.
- Duchen, M.R. Contributions of mitochondria to animal physiology: From homeostatic sensor to calcium signalling and cell death. J. Physiol. 1999, 516, 1–7.
- Halestrap, A.P.; Pasdois, P. The role of the mitochondrial permeability transition pore in heart disease. Biochim. Et Biophys. Acta Bioenerg. 2009, 1787, 1402–1415.
- Duchen, M.R. Roles of mitochondria in health and disease. Diabetes 2004, 53, S96–S102.
- Tang, L.; Gamal El-Din, T.M.; Payandeh, J.; Martinez, G.Q.; Heard, T.M.; Scheuer, T.; Zheng, N.; Catterall, W.A. Structural basis for Ca2+ selectivity of a voltage-gated calcium channel. Nature 2014, 505, 56–61.
- Vafai, S.B.; Mootha, V.K. Mitochondrial disorders as windows into an ancient organelle. Nature 2012, 491, 374–383.
- Pivovarova, N.B.; Andrews, S.B. Calcium-dependent mitochondrial function and dysfunction in neurons. FEBS J. 2010, 277, 3622–3636.
- Moon, H.E.; Paek, S.H. Mitochondrial dysfunction in Parkinson’s disease. Exp. Neurobiol. 2015, 24, 103–118.
- Cullen, P.J.; Lockyer, P.J. Integration of calcium and Ras signalling. Nat. Rev. Mol. Cell Biol. 2002, 3, 339–348.
- Clapham, D.E. Calcium signaling. Cell 2007, 131, 1047–1058.
- Verma, M.; Lizama, B.N.; Chu, C.T. Excitotoxicity, calcium and mitochondria: A triad in synaptic neurodegeneration. Transl. Neurodegener. 2022, 11, 3.
- Khatri, N.; Thakur, M.; Pareek, V.; Kumar, S.; Sharma, S.; Datusalia, A.K. Oxidative stress: Major threat in traumatic brain injury. CNS Neurol. Disord. Drug Targets 2018, 17, 689–695.
- Marjina Singh, A.; Sharma, A.; Narang, R.K.; Singh, G. Management of Hypertension with Conventional and Herbals Drugs. J. Drug Deliv. Ther. 2020, 10, 280–287.
- Joshi, N.J.; Shelke, S.A. Medicinal Plants as Calcium-channel Blockers Against Hypertension. Vascular 2021, 1, 4–5.