Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Othman Al Musaimi | -- | 6506 | 2022-11-03 10:16:54 | | | |
| 2 | Peter Tang | Meta information modification | 6506 | 2022-11-04 02:11:55 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Musaimi, O.A.; Lombardi, L.; Williams, D.R.; Albericio, F. Strategies for Improving Cell-Penetrating Peptides Stability and Delivery. Encyclopedia. Available online: https://encyclopedia.pub/entry/32706 (accessed on 22 July 2026).
Musaimi OA, Lombardi L, Williams DR, Albericio F. Strategies for Improving Cell-Penetrating Peptides Stability and Delivery. Encyclopedia. Available at: https://encyclopedia.pub/entry/32706. Accessed July 22, 2026.
Musaimi, Othman Al, Lucia Lombardi, Daryl R. Williams, Fernando Albericio. "Strategies for Improving Cell-Penetrating Peptides Stability and Delivery" Encyclopedia, https://encyclopedia.pub/entry/32706 (accessed July 22, 2026).
Musaimi, O.A., Lombardi, L., Williams, D.R., & Albericio, F. (2022, November 03). Strategies for Improving Cell-Penetrating Peptides Stability and Delivery. In Encyclopedia. https://encyclopedia.pub/entry/32706
Musaimi, Othman Al, et al. "Strategies for Improving Cell-Penetrating Peptides Stability and Delivery." Encyclopedia. Web. 03 November, 2022.
Copy Citation
Peptides play an important role in many fields, including immunology, medical diagnostics, and drug discovery, due to their high specificity and positive safety profile. However, for their delivery as active pharmaceutical ingredients, delivery vectors, or diagnostic imaging molecules, they suffer from two serious shortcomings: their poor metabolic stability and short half-life. Major research efforts are being invested to tackle those drawbacks, where structural modifications and novel delivery tactics have been developed to boost their ability to reach their targets as fully functional species.
peptides
cell-penetrating peptides
Delivery
1. Introduction
With a molecular weight range from 500 to 5000 Da, peptides occupy an important position between large biological molecules (more than 5000 Da) and the small molecules (less than 500 Da) [1]. Peptides have demonstrated a remarkable performance in various fields of the medical research field. They are deployed in immunology [2], diagnosis [3], drug delivery [1][4][5], as well as biomaterials [6]. They also cover a wide spectrum as active pharmaceutical ingredients (API), carriers, linkers, among others [7]. Their medium size make them more suitable for functions that cannot be accomplished with either small or large molecules. For instance, while the large molecules are incapable of penetrating cells, the hydrophobic interaction is devoid with small molecules [8].
The number of novel therapeutic peptide approvals by the US Food and Drug Administration (USFDA) is increasing on a yearly basis, and currently there are approximately 120 peptides in the market [7][9][10]. About 140 peptide drugs are currently in clinical trials with more than 500 peptides in preclinical trials [7][11][12].
Modern peptide synthesis emerged after the brilliant invention of the Nobel laureate Merrifield in 1963 of solid-phase peptide synthesis (SPPS) [13]. More recently, the introduction of automatic peptide synthesizers, especially microwave-assisted versions, has also greatly facilitated the process whilst reducing the time and effort required for the synthetic process [14]. The implementation of new technologies such as flow chemistry [15] and on line real-time monitoring [16] can significantly optimize reagent usage as well as minimizing reaction time. These developments have facilitated the synthesis of peptides that were difficult to produce using conventional methods.
Despite the biological specificity and the promising safety profile of peptides, they suffer from poor proteolytic stability and short half-lives. These cause a loss of their secondary structure, and thus reduced activity versus time.
Synthetic modifications are one of the possible solutions to enhance the stability of peptides. For instance, swapping L-amino acids with their D-enantiomers provides peptides which are resistant to proteolytic degradation, thereby increasing the half-life [1][17]. However, if the amino acid swapping process compromises the new peptide’s biological activity, then the hydrocarbon stapling or retro inverso strategies are more favorable alternative synthesis tactics [18]. Improved formulations offer an alternate route to deliver the parent peptide without the need to modify it. Figure 1 shows all the strategies covered here for the sake of improving peptide stability and delivery.
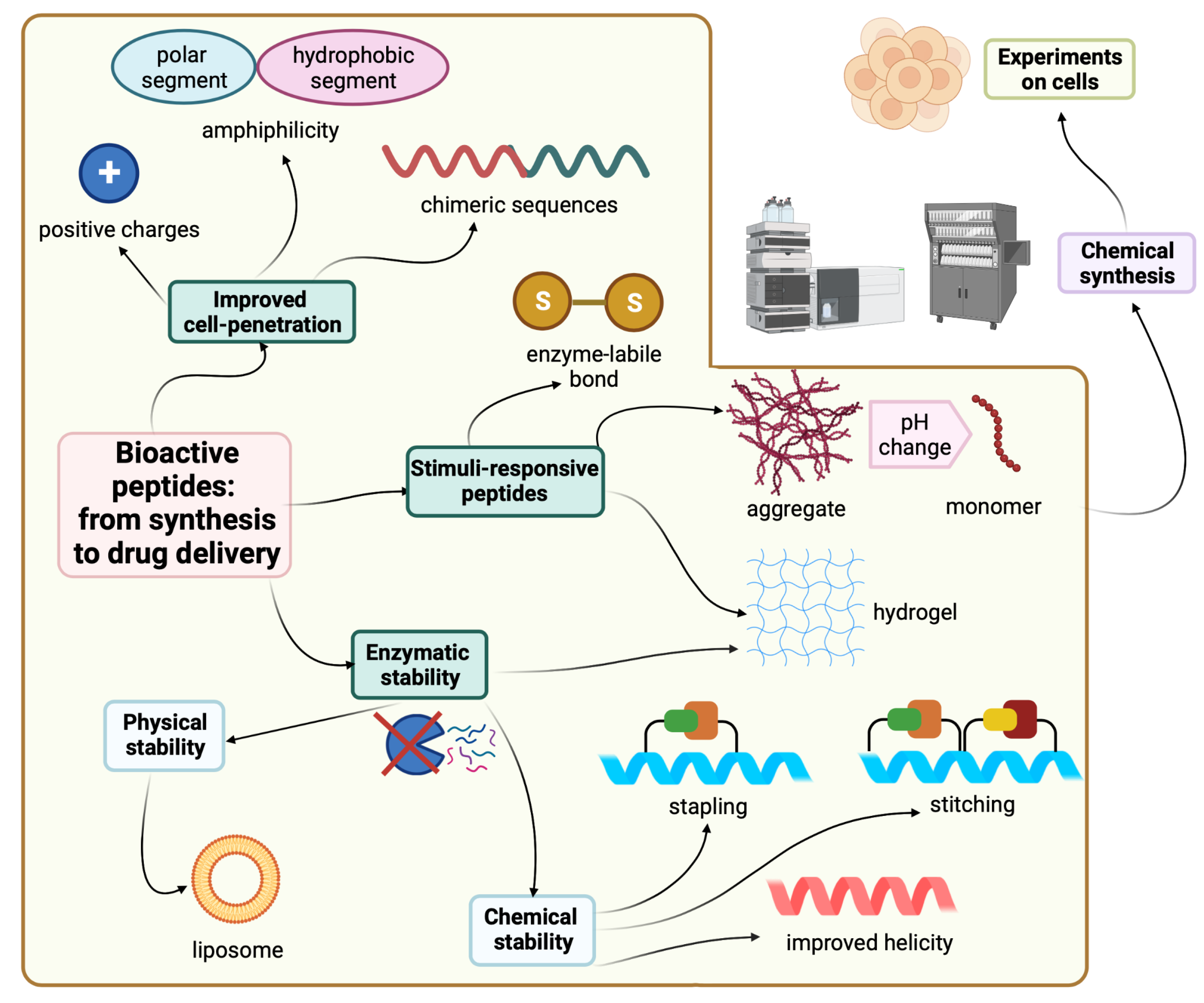
Figure 1. Strategies and tactics to improve peptide stability and delivery. Created with Biorender.com.
2. Cell-Penetrating Peptides (CPPs)
CPPs are a novel class of peptides with a unique cell penetration ability and are also called cell transduction domains (CTDs) [19]. Some diseases and biological processes require biochemical control from within the cell. CPPs can reach specific cellular sites and can facilitate the delivery of associated molecules for therapeutic or imaging purposes [20][21]. CPPs are able to load and deliver different molecules, such as peptides, DNAs, siRNA, and drugs, so they can simply function as delivery vectors [19][20][21][22][23]. The anchoring of the target molecule to the CPP can be done either via covalent or noncovalent electrostatic or hydrophobic interactions [24]. In some cases, the latter is preferred, especially when the covalent attachment might jeopardize the biological activity of the payload [24].
TAT is a regulatory protein expressed from the HIV long terminal repeat (LTR) and it is essential for viral replication. It is composed of 86 amino acid residues comprising two Lys, six Arg in addition, to seven Cys over sixteen residues [25]. Frankel and Pabo noticed that the TAT protein from the human immunodeficiency virus-1 (HIV-1) could be taken by the cells grown in tissue culture and hence transactivate the viral promoter [25].
TAT (GRKKRRQRRRPPQ) is a cell-penetrating peptide identified in a protein component of the HIV-1 virus. The peptide is widely used as a vector for the transport of active molecules into cells and nuclei [26][27]. A shorter TAT peptide sequence has also been shown to have cell-penetrating properties in several studies on cell internalization and gene delivery [28][29]
Green and Loewenstein have synthesized TAT, and a mutant version of it, and demonstrated the fast uptake of the TAT by cells [30]. Interestingly the mutant versions (21 to 41) of the amino acids also showed significant activity. The author suggested that two regions are operational: one for the transactivation and the other for the binding or nuclear targeting region. It is to be mentioned that the aim of all these studies is to study how to disrupt the functionality of the TAT protein to stop the viral replication process that TAT is considered a main part of [30].
Various studies have shown the ability of TAT to translocate through the plasma membrane and then transactivate the viral gene. A fragment of the TAT-derived peptide (37 to 72) was proven to be able to accomplish internalization into various cells and tissues. This fragment comprises basic amino acids which are believed to be responsible for the translocating activity. Furthermore, the fragment exhibits an α-helical structure with amphipathic characteristics. This property could be a key feature for the uptake of this peptide by the enveloped viruses either via fusion or endocytosis mechanisms [30].
Vivès et al. have [31] synthesized several peptide fragments from the shortest sequence that overlaps with two major domains, which are believed to be involved in the cell translocation and nuclear targeting (37–60) of the TAT peptide. The aim of their study was to establish the main domain responsible for translocation and nuclear localization capabilities. The selected sequence has two consecutive Pro residues at its C-terminal located between highly basic residues and the Cys residue. This sequence could act as a spacer between the peptide and the payload, leading to a favorable interaction between the peptide and the cells, resulting in the desired translocation and uptake. Peptide fragments were synthesized, including the selected sequence 37–60, with deletions made at C- and N-termini, as follows: the N-terminal part 38–49 is considered to possess an amphipathic α-helical structure; thus, two fragments were synthesized, 43–60 and 48–60. The C-terminal is considered to be rich with basic amino acids, thus the 37–53 fragment was also synthesized. Interestingly, the 43–60 and 48–60 peptide fragments retained their cell internalization and nuclear accumulation, suggesting the crucial role of the basic domain, as they contain the full basic domain and deletions in the helical domain. On the other hand, the 37–53 fragment was not taken in by the cell even at concentration as high as 20 µM [31].
Surprisingly, the fragments with an α-helical characteristic (37 to 53) seemed not to be crucial, neither for cellular uptake nor the nuclear translocation. On the other hand, all the fragments with basic residues were taken up by cells in less than 5 min with a concentration as low as 100 nM. Furthermore, the study revealed that the helical structure could adversely affect the internalization induced by the basic domain; it slows down the diffusion of the peptide into the cell and towards the nucleus. It is noted that this peptide lacked three Arg residues and Pro-Pro-Gln at the C-terminal, so the authors synthesized another analog to investigate if this defect in the translocation or nuclear targeting is ascribed to the absence of those residues or to the helical structure. The tests confirmed that the absence of those residues had no effect on the reduced efficiency, as no enhancement in the cell uptake or translocation was observed in comparison with the 48–60 fragment. The authors proved that the free sulfhydryl of the Cys residue had no effect on the translocation or localization by comparing the free and the protected sulfhydryl group, and no difference in the functionality was noticed [31].
Wender and coworkers demonstrated the significant role of the (TAT 49–57; RKKRRQRRR) fragment in the translocation process [32]. Further modifications to this sequence, including Ala substitution, resulted in a nonfunctional peptide in terms of cellular uptake. A 9-mer of Arg showed a 20-fold higher cellular uptake with respect to the TAT 49–57, suggesting that the main factor is not the charge itself, but rather the chemical characteristics of the residue itself. Moreover, the 9-mer of the D-Arg showed even a higher uptake rate (100-fold). Obviously, the effect of the guanidium motif is responsible for such effect. This research group designed various polyguanidine-based analogs which revealed an enhanced uptake rate and protease resistance in comparison to either TAT 49–57 or the 9-mer Arg [32].
Park and coworkers confirmed that the Arg and Lys rich sequence (TAT 49–57; RKKRRQRRR) is responsible for the protein transduction through the plasma membrane [33]. Their study showed that any additional deletions at either the C- or N-termini caused a reduction in the transduction capability. The authors fused green fluorescent protein to the previous peptide (TAT 49–57), in addition to nine consecutive Lys residues once and to nine consecutive Arg residues. The rationale behind this selection is the presence of several positively charged residues with the main sequence (six Arg, two Lys, and one Gln), which are believed to have a crucial role in the transduction activity [33].
Interestingly, polylysine (9Lys-GFP) and polyarginine (9Arg-GFP) showed a comparable transduction effect as well as nucleus localization with the main aforementioned sequence. These proteins were successfully delivered to the cell with the same transduction capacity. The importance of the positive charges in the peptide sequence can be ascribed to the interaction between those positive charges and the negative ones on the surface of the cell membrane. In conclusion, this research reaffirms the importance of the basic domain of the TAT peptide for the transduction activity [33].
It is noteworthy that the transduction is also dependent on the characteristic of the fusion target protein. Wender et al. reported the higher transduction efficiency of the 9-mer Arg peptide with respect to TAT 49–57 [32]. This difference is mainly ascribed to the difference in the fusion proteins in both studies. It is to be noted that, in the previous study, the Gly residue at position 48 of the TAT peptide was believed to be part of the nuclear localization signal (NLS). However, as the polyarginine, polylysine, and TAT 49–57 peptides could transduce the GFP and translocate into both the cytoplasm and the nucleus, similar to TAT 48–57 peptide, this clearly refutes the Gly role with respect to NLS [33]. Some studies have stated that the denatured protein containing TAT transduces more efficiently than the correctly folded ones; this behavior could be ascribed to the reduced structural constraints, which enhances the passage of the protein through the cell membrane [34][35][36].
Various mutational analyses of the TAT protein to study its activation mechanism of the human HIV-1 made it possible to propose a functional organization of this protein [25]. In short, the cysteine-rich region of TAT (between 21–38) is important for metal binding, and any sequence mutations in the basic-rich regions would obstruct its functionality [25]. However, studies showed that the metal binding is not required for TAT at the transcription stage [37]. Nevertheless, whilst the activation mechanism of the TAT protein is not fully understood, the binding to the target nucleotide is an important prerequisite for the transactivation to happen [30]. Furthermore, TAT usually accumulates in the nucleus [38]; however, mutating the base rich region of TAT protein (48–52) resulted in a nonfunctional TAT in the cytoplasm [30].
Surprisingly, when other research groups tried to repeat previous studies using shorter versions of the TAT protein, it seems that these selected shorter versions were unable to replicate the performance with the previous group who originated these mutations. Some groups have highlighted the importance of having some periodicity in the acidic, polar, and hydrophobic residues for the TAT to be functional. Loret and coworkers have examined this point by selecting two sequences (2–23 and 38–60) and tried to induce the helicity in them [39].
These sequences were selected as they contain two candidates of activating regions. Circular dichroism (CD) experiments showed that only the 38–60 peptide fragment adopts the α-helical structure in 90% 2,2,2-trifluoroethanol (TFE), whereas in the aqueous solvent, a random-coil structure was obtained. On the other hand, the 2–23 peptide fragments adopted the random-coil structure in both solvents. Molecular dynamic calculations showed that higher temperature caused stretching of the cationic C-terminal of the 38–60 fragment out of the helical structure. The study proved that the 1–14 fragment could be an activating region, not due to the helicity factor, but rather as a Pro-rich region [39].
Peptide transduction domains (PTDs) can facilitate the delivery of potential compounds with promising biological activity in vitro. However, they fail in vivo due to physicochemical constraints, including their size and their lipid solubility. The PTD approach is a smart method which can deliver promising molecules with low or even no intrinsic bioavailability [40].
Comparing the specific PTDs with short functioning sequences suggests that having positively charged amino acids (Arg and Lys) are important for cell permeation and/or interaction with lipids. Those sequences are not structured; they have random coiled structures. However, protein structure prediction algorithms suggests that TAT can adopt helical structures, and the ANTP has a helical structure when present in the homeodomain. As peptides can be transduced even at 4 °C, this means that the absorptive endocytosis is not the mechanism and no receptor is needed. Therefore, any cell can potentially be targeted [35]. Interestingly, when the cells were treated with a drug that inhibits the cellular uptake, such as brefeldin A, the transduction was not affected. Thus, the precise mechanism of the transduction remains a difficult question to answer [35].
The Dowdy group have considered an 11 amino acid fragment of the original TAT peptide (YGRKKRRQRRR) [36]. The transduction occurred rapidly in a concentration-dependent manner and independent of receptors and transporters, and instead is thought to target the lipid bilayer of the cell membrane [36]. The authors investigated the in vivo performance of this fragment by attaching a tetra Gly peptide and a fluorophore (fluorescein isothiocyanate, FITC) FITC-GGGG. It was observed that this motif was quickly transduced into almost all the cultured cells. Next, the FITC-GGGG-11-amino acid TAT peptide was administered into mice along with a control sample of the FITC only. After 20 min, a high-fluorescence signal was obtained in all the blood cells. However, the blood cells of the mice administered with the control sample of FITC showed a slight constant increase in the fluorescence intensity. The TAT-FITC peptide was also transduced in all the splenic cells, the brain cells, as well as skeletal muscle, whereas the control sample remained in the background levels only [36]. This group investigated the possibility of delivering larger biological molecules by fusing a 116 KDa β-galactosidase to the 11 amino acid TAT peptide fragment, resulting in a 120 KDa β-galactosidase-TAT PTD. The control fragment was missing the PTD (11 amino acids part). Both were labeled using FITC. FITC-labeled β-galactosidase-TAT PTD was detected inside the cells and transduced to the maximum intracellular concentrations within less than 15 min, whereas the FITC-labeled β-galactosidase alone was not detected even after 2 h. Injecting these molecules into mice demonstrated the presence of the FITC-labeled β-galactosidase-TAT PTD in all blood and splenic cells, while the FITC-labeled β-galactosidase was not detected. Further successful experiments with other payloads were demonstrated; TAT-Cdk2-DN (human Cdk2 (cyclin-dependent kinase-2) dominant negative) (36 kDa) and TAT-CAK1 (yeast CAK1 (Cdk-activating kinase-1)) (47 kDa) [36].
Joliot et al. synthesized a 60-amino-acid sequence that corresponds to a Drosophila antennapedia complex called homeobox (pAntp) [41]. It is capable of translocating across the neuronal membranes and reaching the nucleus. Isothiocyanate was attached to pAntp to enable the tracing of the pAntp and its penetration ability. A strong fluorescence signal was observed, which suggests that the peptide may enter the cells by nonspecific diffusion or by any uptake mechanism. As pAntp is a basic peptide (pKa 11.35), it could bind to the cell surface and enter only during the course of fixation. The authors investigated this possibility by examining the fluorescent pattern in nonfixed living cells. The results confirmed the internalization of pAntp by live cells. Moreover, after treating the cells with proteinase K, which completely degrades the cell surface molecule neural cell adhesion molecule (NCAM), the fluorescent pAntp was still seen in the nucleus. This confirms that the nuclear localization is not attributed to fixation [41].
Derossit and coworkers investigated several shorter sequences of the antennapedia (60-amino-acid lengths) [42]. In their previous work, they generated a mutant of pAntp called pAntp48S by replacing the Gln at position 50 within the third helix to Ser and deleting two hydrophobic residues (Phe48 and Trp49). In contrast to the wild-type homeodomain, pAntp48S is not internalized by the nerve cells. The authors concluded that the C-terminal region, and specifically that of the third helix, is key for translocation through the plasma membranes. They synthesized various analogs of the C-terminus of pAntp [42]. The authors noticed that the 16-amino-acid sequence derived from the third helix of the homeodomain (RQIKIWFQNRRMKWKK) called pAntp (43–58) as well as the 20 amino acid one are both able to translocate through the membrane at 4 °C, just as with the parent 60-amino-acid one by an energy-independent mechanism. On the other hand, sequences of 15 amino acids are not internalized by cells (Figure 2) [42].

Figure 2. Different sequences. The region that corresponds to the third helix of the homeodomain is shown in blue. The two new F residues that replaced the W are underlined.
The behavior of these peptides was evaluated on 12–22% sodium dodecyl sulfate (SDS)-polyacrylamide. The peptides showed a tendency to aggregate in the presence of SDS and were eluted as dimers. No aggregates were observed in an acidic gel with the absence of SDS, and they were eluted as monomer. The behavior of peptides in a detergent environment could represent that within the lipid bilayer [42].
These peptides at 25 µM were left in contact with the cells for 3.5 h at 37 °C. Peptides 41–60 and 43–58 are internalized where a strong biotin accumulation was observed in the nuclei. Other shorter peptides, 46–60 and 21–55, are not, or are poorly, internalized. To confirm the internalization of 41–60 and 43–58 peptides, freshly dissociated nerve cells were incubated for 2.5 h at 37 °C in a culture medium that contains the peptides at 70 µM. This research confirmed the internalization of both peptides. Moreover, both peptides could be recovered undegraded and with multimeric complexes, reflecting the aggregation enhancement by the presence of lipids. On the contrary, the other two shorter peptides, 46–60 and 41–55, were not present in the cell extracts. Interestingly, the internalization offers a shield against the proteolytic attack, as confirmed by treating the peptides in the cell culture with 0.25% trypsin for 15 min at 37 °C before or after the incubation with cells [42].
Incubating both the 41–60 and 43–58 peptides at low temperature 4 °C showed only little internalization inhibition, whereas the low temperature completely inhibited the little internalization of 46–60 and 41–55 peptides. Lower peptides concentrations were also investigated to understand whether the concentration has an effect on the internalization. Cells were incubated for 2.5 h with various concentrations (from 1.25 to 20 µM). The high concentrations do increase the internalization, whereas the translocation does happen at all concentrations. Peptide 43–58FF, with two Phe residues replaced by two Trp residues at the 48 and 56 positions, showed less internalization efficiency than the original 43–58 peptide analog. Furthermore, 43–58FF showed less tendency to form aggregates, as per the gel analysis [42]. In conclusion, the entire homeodomain crosses the biological membrane more efficiently and its accumulation level is even higher, although it has been administered at lower initial concentration. This behavior could be ascribed to the stronger binding of the entire homeodomain to the target genomic sequence than the shorter peptide versions. The 20- and 16-amino-acid sequences could be recovered intact, suggesting that they were not targeted at the lysosomal compartment. Interestingly, swapping two Trp residues with two Phe diminished the translocating capability. Thus, the internalization is not only attributed to the hydrophobicity only, but it is sequence-dependent as well [42].
The penetratin peptide (RQIKIWFQNRRMKWKK) is another natural CPP. It is derived from a DNA-binding protein in Drosophila fly morphogenesis and has an ability to translocate across neuronal membranes [43]. Liu and coworkers investigated various CPPs for their penetration (RQIKIWFQNRRMKWKK) ability as well as their cellular uptake. The study considers the noninvasive intraocular delivery. Penetratin (derived from a nonviral protein) outranked TAT (GRKKRRQRRRPPQK), 8-mer Arg (R8), low-molecular-weight protamine (VSRRRRRRGGRRRRK), and 8-mer Ser (S8) at both low and high concentrations in the study [44]. The tissue toxicity was carried out by culturing human conjunctival epithelial cells (NHC) with the peptide solution under investigation for 12 h. All the tested peptides showed a low cytotoxic effect at concentrations below 0.3 mM. The lowest tissue toxicity was obtained in the case of penetratin, with an IC50 of higher than 2.5 mM. The most cytotoxic CPP were R8 and protamine, with an average IC50 of 0.7 mM. TAT had an IC50 of 2 mM, which was similar to the control peptide S8, with an IC50 of 2.7 mM. Cellular uptake was determined using fluorescence microscope, where 5-carboxyfluorescein (FAM) was conjugated to the CPPs and a 285 µM concentration FAM-labeled CPPs was considered. No damage or change in the morphology of the NHC cells was observed after treating them with the CPP solutions for 4 h (similar to untreated cells) [44].
The CPPs nonspecifically saturated the cells in a time-dependent manner, with outstanding conjunctiva cell uptake levels being observed for penetratin. TAT was absorbed within 30 min, while protamine and R8 were lower than both penetratin and TAT peptides, but higher than the negative control S8 peptide. Penetratin showed 28- to 153-fold higher cellular uptake than the untreated group and 16- to 29-fold higher cellular uptake than 8-mer Ser (neutral pseudopeptide). The S8 group showed higher (but modest) uptake than the untreated group, and while the group that was treated with the S8 peptide showed a weak fluorescence signal, the groups treated with CPPs showed a much stronger signal, which was localized in the epithelium and sparsely throughout the corneal stroma. The fluorescent pattern was nonspecific and consistent with the cellular uptake data (penetratin > TAT > protamine > R8). After only 10 min, the penetratin reached both the anterior and posterior segments of the eyeballs. The signal reached its peak at 30 min, then started to decrease, but the fluorescent signal was still observed after 6 h. In contrast, only a weak signal was observed in the case of the S8-treated eyes, and the signal diminished within 2 h. CD data showed the S8 had a random-coil conformation, as was the case of protamine and R8, and also for TAT, but to a lesser extent, while for penetratin, its conformation is a type II (PPII) helical structure. The role of the positive charge of the peptide was confirmed, as the neutral control 8-mer Ser pseudopeptide showed weaker permeability with respect to the rest of the examined peptides. However, positive charge was not the only factor that governs permeability. The most efficient peptide in this research, penetratin, was not the most positively charged one. It is worth mentioning that, owing to the amphipathic character of penetratin, it was able to utilize its hydrophilicity while interacting with the continuous secreted tear film, which naturally prevents molecules from getting access to the eye. On the other hand, its lipophilic part was considered to interact with the corneal epithelium. Furthermore, its corneal permeability was 87.5 times more, hitting the threshold of small molecule drugs [44].
In a prestigious study carried out by Patel et al., who compared the capacity for delivering green fluorescent protein (GFP) via different CPPs and their cyclic analogs [45], the following peptides were considered: penetratin, R8, TAT, transportan, and xentry (LCLRPVG). The GFP was selected as a cargo due to its fluorescence property, which can be easily traced and quantified. The cellular uptake was monitored in four different cell lines of different tissue origins (HeLa—human cervical epithelial, HEK—human kidney epithelial, 10 T1/2—mouse embryonic, HepG2—human liver epithelial) [45]. The study revealed that the cellular uptake was dependent on the sequence of the CPP and whether it is linear or cyclized, as well as on the cell line. Interestingly, the special subcellular profiles of the GFP were also different among the different CPPs. HeLa cells were incubated with 10 µM of the CPPs at 37 °C for 1 h. While no CPP was incubated, no fluorescence signal was detected with the confocal microscope. The eGFP-R8 was enriched in perinuclear localization, while the eGFP-transportan was localized towards the periphery of the cells. The eGFP-TAT was similar to that of the eGFP-R8, but with less internalization. As per the flow cytometry analysis, the highest cell uptake of the cationic CPPS (penetratin, R8, and TAT) was for the 10 T1/2 cells. Increased uptake was observed for all CPPS with respect to the internalization by HeLa and HEK cells. The eGFP-transportan was the only peptide to show uptake by the HepG2 cells. The overall performance of CPPs in the different cell lines were as follows: cTAT > cR8 > HA-TAT≈Transportan > R8 > Penetratin ≈ TAT > xentry and 10 T1/2 > HepG2 > HeLa > HEK for the CPPs and for the cell lines tested, respectively. It is worth mentioning that none of the CPPs used caused toxicity in the Hela cells [45]. To investigate the effect of cyclization on the cellular uptake, Cys residue was introduced at both termini in all CPPs (except xentry, which has an endogenous Cys) in order to have the cyclized version of the CPPs form via disulfide bridges. Additional modification was considered to check its effect on the cellular uptake and cytosolic delivery. An endosomal escape sequence (from the influenza virus, HA) was added between the GFP and the TAT peptide. Cyclization and the HA incorporation enhanced the cellular uptake, with the fluorescence signal levels higher in cyclic peptides compared to their linear analogs. Interestingly, the localization was also different in the case of the cyclized GFP-R8 and GFP-TAT with respect to their linear analogs. The cyclization resulted in a more peripheral disposition within the cell. HeLa cells showed the largest uptake in response to cyclized CPP of 4- and 5.9-fold for R8 and TAT, respectively. Furthermore, the addition of HA to the TAT has increased its uptake by all four cell lines. The punctate signal observed in all CPP suggests that endocytosis is involved in the uptake mechanism [45].
The highly cationic nature of the TAT and penetratin peptides is a key to their cell-penetration ability. Indeed, mammalian cell membranes are rich in zwitterionic lipids such as phosphocholines that become polarized in the presence of cationic peptides. This effect is more pronounced with arginine-rich peptides, as they contain guanidinium groups, forming bidentate bonds with phosphate moieties in the lipid molecular structure [46]. Upon interaction with the guanidinium group, arginine-rich cell-penetrating peptides cause the formation of negative Gaussian curvature in the lipid membrane, thus facilitating membrane pore formation [47][48]. Besides natural cell-penetrating peptides, synthetic oligoarginines, such as octa-arginine and cyclic arginine-rich peptides, have been investigated as vehicles for cellular transport [49][50].
Galparan (Transportan) is a chimeric peptide composed of 27 amino acids. It comprises 13 amino acids from the N-terminus of the galanin neuropeptide and 14 amino acids from the C-terminus of the wasp venom peptide toxin (mastoparan) [51]. Mastoparan penetrates the cell membrane and translocates into the inner leaflet, creating short-living pores. Galanin shows various inhibitory effects such as inhibiting the glucose-induced release [51]. Pooga and coworkers have studied the cellular uptake and the localization on the cell surface of the galparan. They synthesized the [Lys13] analog of galparan [52]. Due to high cellular uptake of this peptide, and the available active side chain of the Lys amino acid, which could be used to anchor various cargos to this peptide, the authors coined the name of this novel peptide as Transportan (Figure 3).
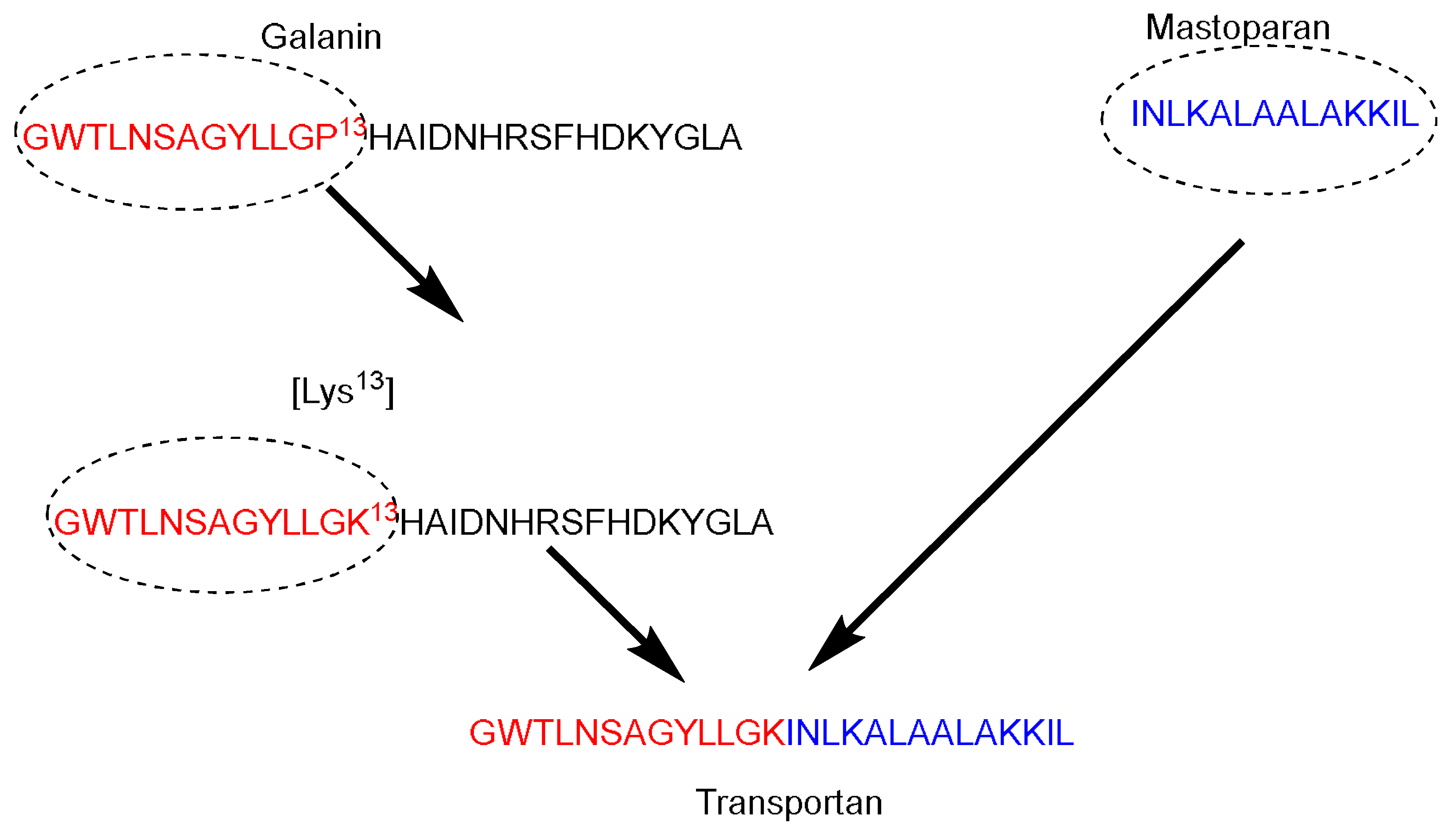
Figure 3. Transportan chimeric peptide sequence. Red: the sequence from galanin neuropeptide; Blue the sequence from the wasp venom peptide toxin (mastoparan).
Nε13-biotinyl-Transportan was shown to be internalized following an energy-independent pathway. The internalization took place efficiently at (37, 4, and 0) °C. In addition, the cellular uptake could not be blocked by treating the cell with various cellular uptake inhibitors [52]. Biotinyl–Transportan was detected throughout the Bowes’s melanoma cells’ interior. Transportan was accumulated in the plasma and nuclear membranes. The peptide was initially internalized quickly at 37 °C. after first 5 min, and it was localized in the plasma membrane and the cytosolic membranous structures (endosomes, endoplasmatic reticulum, and Golgi). There was also some localization in the nucleus, albeit the stain was bright but visible. After 15–30 min, the biotinyl–Transportan was preferentially localized in the nuclear membrane and nuclei. The peptide was also detected at 0 and 4 °C in all membranous structures, but mainly in the plasma membrane. Increasing the incubation time for 1–2 h made the peptide detectable in the nuclear membrane. Thus, at 0 and 4 °C, either the peptide was not penetrating or its concentration was below the detection limit. The penetration feature was noticed in different cell lines as well; however, the authors did not disclose this data [52].
Treating Bowe’s melanoma cells with either hyperosmolar solution of sucrose (which blocks the formation of clathrin-coated pits) or phenylarsine oxide (which crosslinks the thiol groups of membrane surface proteins) did not affect the internalization of biotinyl-transportan. Thus, this confirms that the transportan is not internalized via receptor-mediated endocytosis pathway. It is worth mentioning that after treating Bowe’s melanoma cells with phenylarsine oxide, some cells detached from the surface, however, the remaining cells took the biotinyl–Transportan efficiently. Interestingly, galanin internalization was lost in response to treatment with phenylarsine oxide (the author did not publish this data). The study showed that the internalized biotinyl–Transportan is protected against degradation by trypsin. On the other hand, incubating it with trypsin under the same conditions abolished any uptake, confirming that the trypsin completely digested the biotinyl–Transportan. [52] The 125I-biotinyl-transportan concentration in water was 15-fold higher than in octanol, and independent of concentration. This finding does not contradict its affinity to the membrane, providing that the partition coefficient of 25I-biotinyl-transportan is six-fold more than the 25I-ion from 25INa. Caution must be taken as the octanol/water model does not accurately represent the interaction between the peptide/phospholipids of the membrane. The authors suggested that the internalization of biotinyl–Transportan follows that of antennapedia (inverted micelle mechanism) [42], in which the presence of at least one Trp residue is important. The biotinyl–Transportan has Tyr and Trp residues in its chain, which are important for the micelle formation purposes [52].
A study carried out by Hirose et al. postulated that the peptide penetration is accompanied by the formation of “particle-like” multivesicular structure on the plasma membrane, along with a topical inversion of the plasma membrane [53]. The study used Alexa Fluor 488 conjugated to a 12-mer Arg peptide (R12-Alexa488). Treating cells with the unlabeled peptide, very few species were observed within the plasma membrane, even after using high concentrations. This confirms the role of the Alexa 488 fluorophore as a hydrophobic moiety. Other hydrophobic moieties were also attached to the peptide, confirming these findings. The study showed that attaching R12 peptide to hydrophobic moieties (fluorescent moiety or a peptide tag derived from the human influenza hemagglutinin (HAtag YPYDVPDYA)) helps in stimulating certain dynamic morphological alterations in the plasma membrane that allow the peptide to permeate through the plasma membrane [53].
This research was based on the hypothesis that the peptide diffusion happens at specific sites of the plasma membrane. The penetration of the peptide into the cell happened through specific sites, as confirmed by strong florescent signals, and then spread to the rest of the plasma membrane. Differential interference contrast microscopy revealed that the internalization of the peptide was accompanied by the formation of a particle-like structure. The locations of these particles were matching with the sites of peptide infusion. As the R4-Alexa488 peptide had poor translocation ability, no significant alterations in the plasma membrane were observed. The research confirmed the occurrence of the translocation even at 4 and 15 °C, which means that the translocation is an energy-independent process, provided that the addition of macropinocytosis inhibitors did not affect the particle-like formation [53]. The authors studied the effect of the membrane potential on the particle formation. The high Na+ concentration is considered a normal condition that maintains the membrane potential (physiological condition). On the other hand, having K+ in high concentrations instead diminishes the membrane potential. Using an Na+-rich buffer to incubate the cells showed the expected particle-like formation, whereas this was not the case when having the K+ as predominant in the extracellular medium, even when high concentrations of the R12-Alexa488 were considered. Interestingly, treating the cells with an excess of Na+ after they were treated with an excess of K+ showed a recovery in the influx of the peptide [53].
In summary, the interaction of the peptide with the plasma membrane is also dependent on the membrane potential and not only the peptide itself. Membrane inversion was also accompanied in the influx process. Phosphatidylserine is a membrane component that is localized to the inner side of the plasma membrane at the normal conditions. On the other hand, under certain conditions, it could be interrupted, such as in the case of apoptosis, in which it would be inverted, which could be investigated using specific binding partner. Such membrane movement (inversion) was observed with the R12-Alexa488 and not with the R4-Alexa488 one [53].
The research highlighted some dynamic rearrangements of the membrane with the interacting molecules. For example, the acidic constituents interacted with the basic residues of the R12. Scanning electron microscope (SEM) analysis of the plasma membrane upon penetration of R12-Alexa488 revealed the presence of various small vesicles instead of a single large one. However, eventually the accumulation of the peptide will lead to the formation of large particles at the site of influx. Such observation was not seen in the control cells (untreated cells). Transmission electron microscope (TEM) analysis confirmed the formation of the particles at both low and physiological temperature (4 and 37) °C, respectively. This work confirms that the structural alterations in the plasma membrane are endocytosis-independent and happened as a result of the physicochemical interactions between the peptide and the plasma membrane [53]. Interestingly, as the study revealed no leakage of the lactate dehydrogenase from the treated cells, this means that the plasma membrane retained its integrity. Several membrane-repair mechanisms are operating at such conditions. However, the authors could not determine which was operational here. However, they confirmed that neither lysosome nor endosome-mediated membrane-repair was taking place. R4-Alexa488 has low permeability and thus no significant alterations in the plasma membrane. On the other hand, treating HeLa cells with this peptide in the presence of unlabeled R4, R12, and R12-HAtag led to a noticeable increase in the cellular uptake of R4-Alexa488, in addition to membrane-particle formation. Treating the cells with R4-Alexa488 peptide with unlabeled R12 showed only little increase in the cellular uptake. The addition of unlabeled R4 did not increase the cellular uptake of R4-Alexa488 [53]. The authors checked whether the particle formation was due to the simple interactions between the peptide with the lipids or not. They studied the R12-Alexa488 with giant vesicles that mimic the composition of the plasma membrane. No peptide translocation was observed, confirming that the particles formed are not due to a simple interaction between peptides with lipids [53].
El-Andaloussi and coworkers have studied the delivery capability of M918. M918 is a 22 AAs peptide (MVTVLFRRLRIRRACGPPRVRV-NH2) derived from the tumor-suppressor protein p14ARF [54]. It comprises the fragments from 1–22, with inverted positions for 3–8, with seven positive charges. Initially the peptide was prepared as a control to mimic the activity of the ARF protein. Surprisingly, it showed an excellent penetration capability [54]. These authors investigated the following five CPPs versus the M918: penetratin, transportan 10 (TP10, AGYLLGK*INLKALAALAKKIL-NH2) a 21 AAs that represents a deletion analog of Transportan, YDEGE, M705 PNA (CysKKCCTCTTACCTCAGTTACAKK+-NH2), and M705 inverted PNA (CysKKCCTCTTACACTCGTTACAKK+-NH2) [54]. The results showed the better internalization of M918 than penetratin and TP10 f HeLa cells and Chinese hamster ovarian (CHO) cells. YDEGE was used as a negative control (it showed negligible uptake) and to roll out any uptake effect as a result of membrane disruption. Moreover, no lactate dehydrogenase (LDH) leakage was observed, except in the case of TP10, where a massive membrane leakage was observed at 25 µM. M918 did not show any cell proliferation after 72 h with any of the cell lines under investigation. As the high cellular uptake could be ascribed to the peptide aggregation in the cellular membrane, the authors analyzed the uptake using the confocal microscope. No clusters of the peptide were observed in breast cancer MCF-7 cells or HeLa cells. Some membrane-associated peptides were observed in astrocytoma cells, Hifko. The majority of the internalized materials reside in the vesicles, and a negligible amount in the cell nucleus. M918 was further studied and checked for its delivery potentials [54]. Avidin and streptavidin were chosen as model proteins for the transduction experiments. Physical mixtures of streptavidin or avidin with penetratin, TP10, and M918 peptides (noncovalent complex) proved that the three tested peptides were able to promote the cellular uptake of fluorescein-labeled streptavidin at a 5 µM concentration, with the M918 being superior to penetratin and TP10 [54]. The same peptides were tested at a 1 µM concentration; however, with no enhancement in the delivery. On the other hand, with 1 µM of the biotinylated peptides, a noticeable uptake enhancement was observed, in particular with TP10 and M918. Penetratin was low in both strategies. The study proved the superiority of M918 to deliver antisense PNA-targeting pre-mRNA over TP10 and penetratin. Confocal microscope study confirmed that the distribution of the peptide is within the cells and no clusters were observed on the cellular membranes. Therefore, the high cellular uptake is attributed to the internalized peptide rather than to the aggregation in the cellular membranes [54]. Cells were treated and incubated with M918 at 4 °C. The cellular uptake was inhibited at low temperature (4 °C) in the HeLa cells. The same was noticed in the CHO cells when treated with penetratin and TP10. The addition of endocytosis inhibitors, cytochalasin D, which blocks the actin polymerization that is important for the formation of micropinosomes and wortmannin, which is a PI3-kinase inhibitor that also inhibits clathrin-mediated endocytosis, resulted in a decreased uptake of M918. Thus, the authors concluded that the endocytosis pathway (macropinocytosis) mediated the uptake, at least partially. Some studies emphasized the necessity of glycosaminoglycans (GAGs) on the cell surface for the uptake process. While using the GAG-deficient CHO cell, a significant decrease in the uptake of TP10 and penetratin was observed with respect to the wild-type CHO cells. On the other hand, only a little decrease in the uptake of M918 was observed when the cells were treated with the three peptides which were covalently linked to fluoresceinyl-labeled PNA (via a disulfide bridge). The luciferase was measured after 16 h considering 5 µM concentrations of peptides [54].
M918 proved to be the most potent vector, where a 15-fold increase in the splicing was observed versus 10- and 5-fold for TP10 and penetratin, respectively, with respect to the untreated cells. Most importantly, neither inverted PNA conjugated to M918 nor naked PNA induced significant splicing at 10 µM, which proves the importance of the CPPs for cellular uptake. As the endosomal entrapment is a major concern for the endocytosis pathways, the authors were able to circumvent this concern by adding a lysosomotrophic agent (chloroquine), and the splicing was improved with respect to M918-PNA without the chloroquine [54].
References
- Al Musaimi, O.; Al Shaer, D.; De la Torre, B.G.; Albericio, F. 2017 FDA Peptide Harvest. Pharmaceuticals 2018, 11, 42.
- Jensen, S.M.; Potts, G.K.; Ready, D.B.; Patterson, M.J. Specific MHC-I Peptides Are Induced Using PROTACs. Front. Immunol. 2018, 9, 2697.
- Jeong, W.J.; Bu, J.; Kubiatowicz, L.J.; Chen, S.S.; Kim, Y.; Hong, S. Peptide-nanoparticle conjugates: A next generation of diagnostic and therapeutic platforms? Nano Converg. 2018, 5, 38.
- Al Shaer, D.; Al Musaimi, O.; Albericio, F.; de la Torre, B.G. 2018 FDA Tides Harvest. Pharmaceuticals 2019, 12, 52.
- Du, A.W.; Stenzel, M.H. Drug carriers for the delivery of therapeutic peptides. Biomacromolecules 2014, 15, 1097–1114.
- Tsoras, A.N.; Champion, J.A. Protein and Peptide Biomaterials for Engineered Subunit Vaccines and Immunotherapeutic Applications. Annu. Rev. Chem. Biomol. Eng. 2019, 10, 337–359.
- Al Shaer, D.; Al Musaimi, O.; Albericio, F.; de la Torre, B.G. 2021 FDA TIDES (Peptides and Oligonucleotides) Harvest. Pharmaceuticals 2022, 15, 222.
- Hopkins, A.L.; Groom, C.R. The druggable genome. Nat. Rev. Drug Discov. 2002, 1, 727–730.
- D’Aloisio, V.; Dognini, P.; Hutcheon, G.A.; Coxon, C.R. PepTherDia: Database and structural composition analysis of approved peptide therapeutics and diagnostics. Drug Discov. 2021, 26, 1409–1419.
- Muttenthaler, M.; King, G.F.; Adams, D.J.; Alewood, P.F. Trends in peptide drug discovery. Nat. Rev. Drug Discov. 2021, 20, 309–325.
- Fosgerau, K.; Hoffmann, T. Peptide therapeutics: Current status and future directions. Drug Discov. 2015, 20, 122–128.
- Lau, J.L.; Dunn, M.K. Therapeutic peptides: Historical perspectives, current development trends, and future directions. Bioorg. Med. Chem. 2018, 26, 2700–2707.
- Merrifield, R.B. Solid Phase Peptide Synthesis. I. The Synthesis of a Tetrapeptide. J. Am. Chem. Soc. 1963, 85, 2149–2154.
- Collins, J.M.; Porter, K.A.; Singh, S.K.; Vanier, G.S. High-efficiency solid phase peptide synthesis (HE-SPPS). Org. Lett. 2014, 16, 940–943.
- Mijalis, A.J.; Thomas, D.A., 3rd; Simon, M.D.; Adamo, A.; Beaumont, R.; Jensen, K.F.; Pentelute, B.L. A fully automated flow-based approach for accelerated peptide synthesis. Nat. Chem. Biol. 2017, 13, 464–466.
- Sletten, E.T.; Nuno, M.; Guthrie, D.; Seeberger, P.H. Real-time monitoring of solid-phase peptide synthesis using a variable bed flow reactor. Chem. Commun. 2019, 55, 14598–14601.
- Brayden, D.J.; Hill, T.A.; Fairlie, D.P.; Maher, S.; Mrsny, R.J. Systemic delivery of peptides by the oral route: Formulation and medicinal chemistry approaches. Adv. Drug. Deliv. Rev. 2020, 157, 2–36.
- Chorev, M. The partial retro–inverso modification: A road traveled together. Biopolymers 2005, 80, 67–84.
- Derakhshankhah, H.; Jafari, S. Cell penetrating peptides: A concise review with emphasis on biomedical applications. Biomed. Pharmacother. 2018, 108, 1090–1096.
- Stiltner, J.; McCandless, K.; Zahid, M. Cell-Penetrating Peptides: Applications in Tumor Diagnosis and Therapeutics. Pharmaceutics 2021, 13, 890.
- Khan, M.M.; Filipczak, N.; Torchilin, V.P. Cell penetrating peptides: A versatile vector for co-delivery of drug and genes in cancer. J. Control. Release 2021, 330, 1220–1228.
- Desale, K.; Kuche, K.; Jain, S. Cell-penetrating peptides (CPPs): An overview of applications for improving the potential of nanotherapeutics. Biomater. Sci. 2021, 9, 1153–1188.
- Falanga, A.; Lombardi, L.; Galdiero, E.; Genio, V.D.; Galdiero, S. The world of cell penetrating: The future of medical applications. Future Med. Chem. 2020, 12, 1431–1446.
- Xie, J.; Bi, Y.; Zhang, H.; Dong, S.; Teng, L.; Lee, R.J.; Yang, Z. Cell-Penetrating Peptides in Diagnosis and Treatment of Human Diseases: From Preclinical Research to Clinical Application. Front. Pharmacol. 2020, 11, 697.
- Frankel, A.D.; Pabo, C.O. Cellular uptake of the tat protein from human immunodeficiency virus. Cell 1988, 55, 1189–1193.
- Chugh, A.; Eudes, F. Translocation and nuclear accumulation of monomer and dimer of HIV-1 Tat basic domain in triticale mesophyll protoplasts. Biochim. Biophys. Acta BBA Biomembr. 2007, 1768, 419–426.
- Schwarze, S.R.; Hruska, K.A.; Dowdy, S.F. Protein transduction: Unrestricted delivery into all cells? Trends Cell Biol. 2000, 10, 290–295.
- Brooks, N.A.; Pouniotis, D.S.; Tang, C.-K.; Apostolopoulos, V.; Pietersz, G.A. Cell-penetrating peptides: Application in vaccine delivery. Biochim. Biophys. Acta Rev. Cancer 2010, 1805, 25–34.
- Lakshmanan, M.; Kodama, Y.; Yoshizumi, T.; Sudesh, K.; Numata, K. Rapid and Efficient Gene Delivery into Plant Cells Using Designed Peptide Carriers. Biomacromolecules 2013, 14, 10–16.
- Green, M.; Loewenstein, P.M. Autonomous functional domains of chemically synthesized human immunodeficiency virus tat trans-activator protein. Cell 1988, 55, 1179–1188.
- Vivès, E.; Brodin, P.; Lebleu, B. A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J. Biol. Chem. 1997, 272, 16010–16017.
- Wender, P.A.; Mitchell, D.J.; Pattabiraman, K.; Pelkey, E.T.; Steinman, L.; Rothbard, J.B. The design, synthesis, and evaluation of molecules that enable or enhance cellular uptake: Peptoid molecular transporters. Proc. Natl. Acad. Sci. USA 2000, 97, 13003–13008.
- Park, J.; Ryu, J.; Kim, K.A.; Lee, H.J.; Bahn, J.H.; Han, K.; Choi, E.Y.; Lee, K.S.; Kwon, H.Y.; Choi, S.Y. Mutational analysis of a human immunodeficiency virus type 1 Tat protein transduction domain which is required for delivery of an exogenous protein into mammalian cells. J. Gen. Virol. 2002, 83 Pt 5, 1173–1181.
- Kwon, H.Y.; Eum, W.S.; Jang, H.W.; Kang, J.H.; Ryu, J.; Ryong Lee, B.; Jin, L.H.; Park, J.; Choi, S.Y. Transduction of Cu,Zn-superoxide dismutase mediated by an HIV-1 Tat protein basic domain into mammalian cells. FEBS Lett. 2000, 485, 163–167.
- Nagahara, H.; Vocero-Akbani, A.M.; Snyder, E.L.; Ho, A.; Latham, D.G.; Lissy, N.A.; Becker-Hapak, M.; Ezhevsky, S.A.; Dowdy, S.F. Transduction of full-length TAT fusion proteins into mammalian cells: TAT-p27Kip1 induces cell migration. Nat. Med. 1998, 4, 1449–1452.
- Schwarze, S.R.; Ho, A.; Vocero-Akbani, A.; Dowdy, S.F. In vivo protein transduction: Delivery of a biologically active protein into the mouse. Science 1999, 285, 1569–1572.
- Jeyapaul, J.; Reddy, M.R.; Khan, S.A. Activity of synthetic tat peptides in human immunodeficiency virus type 1 long terminal repeat-promoted transcription in a cell-free system. Proc. Natl. Acad. Sci. USA 1990, 87, 7030–7034.
- Malim, M.H.; Hauber, J.; Le, S.Y.; Maizel, J.V.; Cullen, B.R. The HIV-1 rev trans-activator acts through a structured target sequence to activate nuclear export of unspliced viral mRNA. Nature 1989, 338, 254–257.
- Loret, E.P.; Vives, E.; Ho, P.S.; Rochat, H.; Van Rietschoten, J.; Johnson, W.C., Jr. Activating region of HIV-1 Tat protein: Vacuum UV circular dichroism and energy minimization. Biochemistry 1991, 30, 6013–6023.
- Schwarze, S.R.; Dowdy, S.F. In vivo protein transduction: Intracellular delivery of biologically active proteins, compounds and DNA. Trends Pharmacol. Sci. 2000, 21, 45–48.
- Joliot, A.; Pernelle, C.; Deagostini-Bazin, H.; Prochiantz, A. Antennapedia homeobox peptide regulates neural morphogenesis. Proc. Natl. Acad. Sci. USA 1991, 88, 1864–1868.
- Derossi, D.; Joliot, A.H.; Chassaing, G.; Prochiantz, A. The third helix of the Antennapedia homeodomain translocates through biological membranes. J. Biol. Chem. 1994, 269, 10444–10450.
- Derossi, D.; Chassaing, G.; Prochiantz, A. Trojan peptides: The penetratin system for intracellular delivery. Trends Cell Biol. 1998, 8, 84–87.
- Liu, C.; Tai, L.; Zhang, W.; Wei, G.; Pan, W.; Lu, W. Penetratin, a potentially powerful absorption enhancer for noninvasive intraocular drug delivery. Mol. Pharm. 2014, 11, 1218–1227.
- Patel, S.G.; Sayers, E.J.; He, L.; Narayan, R.; Williams, T.L.; Mills, E.M.; Allemann, R.K.; Luk, L.Y.P.; Jones, A.T.; Tsai, Y.-H. Cell-penetrating peptide sequence and modification dependent uptake and subcellular distribution of green florescent protein in different cell lines. Sci. Rep. 2019, 9, 6298.
- Stanzl, E.G.; Trantow, B.M.; Vargas, J.R.; Wender, P.A. Fifteen years of cell-penetrating, guanidinium-rich molecular transporters: Basic science, research tools, and clinical applications. Acc. Chem. Res. 2013, 46, 2944–2954.
- Chan, D.I.; Prenner, E.J.; Vogel, H.J. Tryptophan-and arginine-rich antimicrobial peptides: Structures and mechanisms of action. Biochim. Biophys. Acta BBA Biomembr. 2006, 1758, 1184–1202.
- Schmidt, N.; Mishra, A.; Lai, G.H.; Wong, G.C. Arginine-rich cell-penetrating peptides. FEBS Lett. 2010, 584, 1806–1813.
- Nakase, I.; Niwa, M.; Takeuchi, T.; Sonomura, K.; Kawabata, N.; Koike, Y.; Takehashi, M.; Tanaka, S.; Ueda, K.; Simpson, J.C.; et al. Cellular uptake of arginine-rich peptides: Roles for macropinocytosis and actin rearrangement. Mol. Ther. 2004, 10, 1011–1022.
- Mandal, D.; Nasrolahi Shirazi, A.; Parang, K. Cell-penetrating homochiral cyclic peptides as nuclear-targeting molecular transporters. Angew. Chem. Int. Ed. 2011, 50, 9633–9637.
- Langel, Ü.; Pooga, M.; Kairane, C.; Zilmer, M.; Bartfai, T. A galanin-mastoparan chimeric peptide activates the Na+,K(+)-ATPase and reverses its inhibition by ouabain. Regul. Pept. 1996, 62, 47–52.
- Pooga, M.; Hällbrink, M.; Zorko, M.; Langel, Ü. Cell penetration by transportan. FASEB J. 1998, 12, 67–77.
- Hirose, H.; Takeuchi, T.; Osakada, H.; Pujals, S.; Katayama, S.; Nakase, I.; Kobayashi, S.; Haraguchi, T.; Futaki, S. Transient focal membrane deformation induced by arginine-rich peptides leads to their direct penetration into cells. Mol. Ther. 2012, 20, 984–993.
- El-Andaloussi, S.; Johansson, H.J.; Holm, T.; Langel, Ü. A novel cell-penetrating peptide, M918, for efficient delivery of proteins and peptide nucleic acids. Mol. Ther. 2007, 15, 1820–1826.
More
Information
Subjects:
Chemistry, Medicinal
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.7K
Entry Collection:
Peptides for Health Benefits
Revisions:
2 times
(View History)
Update Date:
04 Nov 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No