Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | August Wrotek | -- | 4508 | 2022-10-31 13:11:50 | | | |
| 2 | Vivi Li | + 23 word(s) | 4531 | 2022-11-01 02:33:48 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Wrotek, A.; Jackowska, T. Respiratory Syncytial Virus and Air Pollution Interaction. Encyclopedia. Available online: https://encyclopedia.pub/entry/32110 (accessed on 08 August 2026).
Wrotek A, Jackowska T. Respiratory Syncytial Virus and Air Pollution Interaction. Encyclopedia. Available at: https://encyclopedia.pub/entry/32110. Accessed August 08, 2026.
Wrotek, August, Teresa Jackowska. "Respiratory Syncytial Virus and Air Pollution Interaction" Encyclopedia, https://encyclopedia.pub/entry/32110 (accessed August 08, 2026).
Wrotek, A., & Jackowska, T. (2022, October 31). Respiratory Syncytial Virus and Air Pollution Interaction. In Encyclopedia. https://encyclopedia.pub/entry/32110
Wrotek, August and Teresa Jackowska. "Respiratory Syncytial Virus and Air Pollution Interaction." Encyclopedia. Web. 31 October, 2022.
Copy Citation
Respiratory syncytial virus (RSV) is one of the major infectious agents in paediatrics, and its relationship with air pollution is frequently observed. A relationship between lower air quality and RSV infections was observed mainly in hospital-based and some community-based studies, with particular interest in PM2.5, PM10, nitric dioxide (NO2), ozone, carbon monoxide (CO), and benzene. Exposure to another pollutant, cigarette smoke, is a well-described risk factor for a more severe RSV infection in children, an increased RSV LRTI prevalence, and chronic obstructive pulmonary disease (COPD); in the latter case, the RSV can be persistently present, escaping an immune response.
respiratory syncytial virus
air pollution
particulate matter
PM2.5
pathomechanism
environmental pollutants
bronchial hyperreactivity
1. Introduction
Air pollution is one of the major public health concerns due to a well-established and still growing body of evidence on its detrimental effects on human health; according to the World Health Organization (WHO), air pollution is estimated to contributs to approximately 7 million premature deaths globally [1][2]. Air pollutants may be classified depending on their 1. Formation—primary (emitted directly) versus secondary (formed as a result of reactions with other pollutants or gases), 2. Origin—indoor versus outdoor, or 3. physical characteristics—gaseous (nitrogen oxides, NOx, sulfur dioxide, SO2, ozone, O3, carbon monoxide, CO, specific volatile organic compounds, SVOC (e.g., benzene) versus particulate (particulate matter smaller than or equal to 10 µm in diameter, PM10 or coarse PM; particulate matter between 0.1 and 2.5 µm in diameter, PM2.5 or fine PM; particulate matter smaller than or equal to 0.1 µm in diameter, ultrafine PM) [3]. The studies on the influence of air pollution on human health have mainly revealed its relationship with cardiovascular and respiratory diseases; however, outdoor air pollution and particulate matter are also listed by the WHO agency and the International Agency for Research on Cancer (IARC) as carcinogenic hazards to humans [4][5]. Thus, the WHO announced that the six most significant air pollutants include particulate matter smaller than or equal to 2.5 µm in diameter (PM2.5), particulate matter smaller than or equal to 10 µm in diameter (PM10), ozone (O₃), nitrogen dioxide (NO₂), sulfur dioxide (SO₂), and carbon monoxide (CO), and the updates on the highest tolerable levels of those pollutants are published in WHO Global Air Quality Guidelines (AQGs) on a regular basis [1].
Respiratory syncytial virus (RSV) is one of the most significant etiological agents of respiratory tract infections (RTI), both due to its burden, as well as clinical severity; the global estimates report RSV as the cause of 33 million lower RTI (LRTI) episodes and 3.6 million of hospitalizations per year, and those numbers are restricted to children under 5 years of age only [6]. Two percent of deaths in children under 5 years of age, and 3.6% in children aged 1–6 months are related to RSV [6]. Moreover, RSV, although initially considered to be a virus of infancy only, turned out to be one of the major infective factors in adults, especially in the elderly [7][8][9]; despite a limited access to viral testing and often unspecific clinical picture of RSV RTI, the awareness of the significance of RSV is growing [10]. The high frequency and severity of the RSV disease translate into a huge socioeconomic impact and utilization of healthcare resources seen in paediatric and adult population [7][11][12][13], urging a development of monoclonal antibodies and vaccines targeting infants, pregnant women and older adults [14]. A relationship between lower air quality and RSV infections was observed mainly in hospital-based and some community-based studies [15][16][17][18][19][20][21], with particular interest in PM2.5 [17][18][21], PM10 [15][21], nitric dioxide (NO2) [21], ozone [22], carbon monoxide (CO) [20], and benzene [19]. Exposure to another pollutant, cigarette smoke, is a well-described risk factor for a more severe RSV infection in children, an increased RSV LRTI prevalence [23][24][25], and chronic obstructive pulmonary disease (COPD) [26]; in the latter case, the RSV can be persistently present, escaping an immune response [27].
Air pollutants induce a complex interaction via various pathogenetic pathways in the respiratory tract, including an increased production of reactive oxygen species (ROS), an activation of transcription factors, or the production of cytokines and/or chemokines; as a result, local homeostasis is disturbed, lung immunity is reduced, inflammatory response is exacerbated, and hyperreactivity is increased [28]. While some of the mechanisms are common and expected to be repeatedly present in a response to various etiological factors, the other ones might be pathogen-specific [28]. Nonetheless, an in-depth knowledge on the molecular bases of the air pollutants–RSV interaction is indispensable for planning any wide-spread action counteracting the effects of air pollution. Moreover, when a specific mechanism is identified, targeted actions, including pharmacological treatment, might be considered.
2. Molecular Mechanisms
2.1. A Facilitated Viral Entry
2.1.1. Epithelial Barrier
Epithelium forms a first-line barrier in the airways that is crucial for antiviral defense, and a damage to the airway epithelium is characteristic for an RSV infection [29][30]. The RSV has been previously shown to cause a disintegration of the epithelium and its increased permeability (so called “leaky epithelium”), mainly due to a disruption of the apical junctional complexes (AJC), which are intercellular complexes [30]. Except for a diminished integrity, the RSV also causes a remodeling of the actin cytoskeletal to which AJC is linked [29][30]. The RSV-induced epithelial disintegration might be enhanced by an exposure to nanoparticles and titanium-dioxide nanoparticles (TiO2-NP), which was shown in a number of studies to be a suitable model for the studies on pulmonary effects of environmental nanoparticles (see Figure 1) [29][31][32][33][34]. Both in vitro (immortalized human bronchial epithelial cells) and in vivo (murine) models of TiO2-NP effects, investigated by Smallcombe, confirmed the hypothesis of an augmented barrier dysfunction [29]. The barrier dysfunction comprised an amplified AJC disruption and led to an increased viral replication [29]. The molecular mechanism of the epithelium integrity disruption by TiO2-NP was shown to be related to an oxidative stress and a generation of reactive oxygen species (ROS) and a pretreatment with an antioxidant (N-acetylcysteine) not only attenuated the AJC disruption, but was also able to reverse the enhanced RSV infection [29].
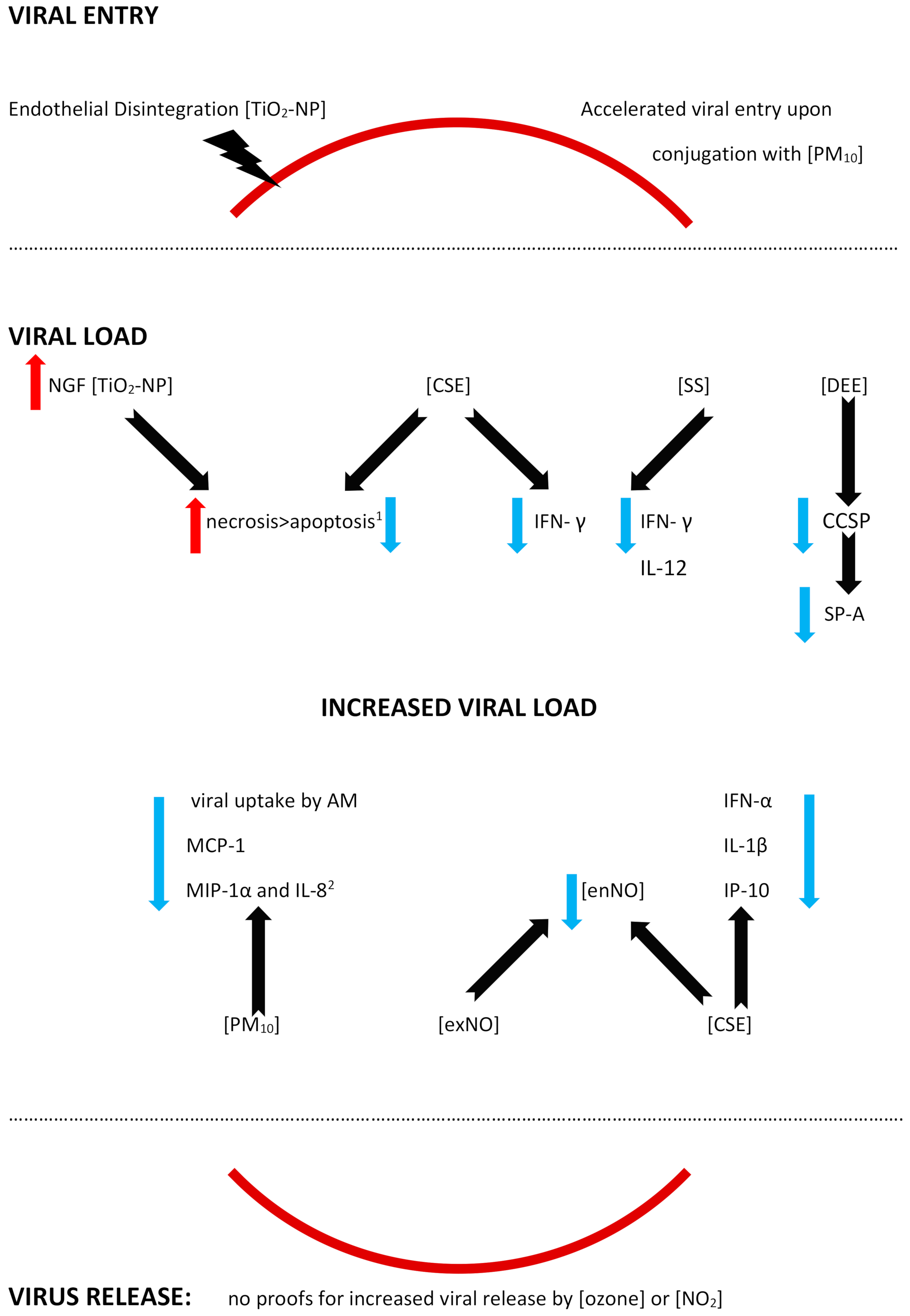
Figure 1. A diagram of the feasible mechanisms facilitating an RSV infection upon an exposure to the different air pollutants. The activity of the air pollutants is shown with the black solid arrow, while the directions of the effects are marked with red (increase) or blue (decrease) arrows; the red solid line symbolizes a division between the extracellular and intracellular space (in the case of a viral entry and a viral release), and the red solid lines are separated from the rest of the figure by black dotted lines, since the remaining effects (shown in the central part of the scheme) are reported in extra- or intracellular spaces. Please note: the figure has only a demonstrative character and is based on the literature found for the purposes of this entry. The effects of the air pollutants might vary with regards to the presence/absence of the mechanism, its extent, and its effects. The mechanisms might be influenced by other, not verified or not shown, pathways. The mechanisms might be dose-dependent, time-dependent, exposition sequence-dependent, and model-dependent (i.e., differences between the models used in the studies and human beings might be seen). The figure is simplified, and, for example, the viral load here is a common understanding of the enhanced presence of an RSV load, measured in different ways, and depending on the study (increased expression of the RSV genes and/or proteins, viral titers, reappearance of replication, etc.); for details, see the text and/or refer to the original articles. In addition, contradictory effects of the same air pollutant might be reported, depending on the study model. Abbreviations: CSE—cigarette smoke extract, DEE—diesel engine emissions, NO2—nitrogen dioxide, PM10—particulate matter smaller than or equal to 10 µm in diameter, SS—s ide-stream cigarette smoke, TiO2-NP—titanium-dioxide nanoparticles; AM—alveolar macrophage, CCSP—Clara cell secretory protein, IL-(1β, 8, 12)—Interleukin-(1β, 8, 12), IFN-α—interferon α, IFN-γ—Interferon γ, IP-10- IFN—γ-inducible protein (=CXCL 10), MCP-1—monocyte chemoattractant protein-1 (=CCL2), MIP-1α—macrophage inflammatory protein-1 α (=CCL3), ex/enNO—exogenous/endogenous nitrogen oxide, and SP-A—surfactant protein A. 1—increased cell death (probably due to necrosis) and a decreased apoptosis in the case of cigarette smoke extract, and 2—a lack of an additive effect, which may be interpreted as a relative decrease.
2.1.2. A Facilitated Viral Entry
A facilitated viral entry was reported in an in vitro mimic of PM10 containing the RSV that were deposited onto the airway epithelial cells [35]. As a result, an accelerated viral entry was observed upon the RSV conjugation to PM, and the mechanism involved an endocytic pathway [35]. Moreover, the RSV survival was increased when it was associated with PM10 in the model [35]. No studies proving enhanced viral receptors expression on the respiratory tract cells were found.
2.2. An Altered Viral Load
- (A)
-
An Increased Viral Load
An increased viral load, increased RSV gene and/or protein expression, or its replication outside the epidemic season was reported after exposure to various air pollutants, such as cigarette smoke, particulate matter, nanoparticles, or nitric oxide [31][36][37][38][39][40][41]. The studies on the mechanisms underlying an increase in infections report different pathways, including enhanced necrosis and decreased apoptosis, a mitigated antiviral defense via altered functions of the epithelium, alveolar macrophages or dendritic cells, or a decreased inflammatory response [31][36][37][38][39][40][41].
2.2.1. Autophagy, Decreased Apoptosis, and Enhanced Necrosis
Autophagy is crucial for the immune response, since the structures of damaged cells might be cut into recyclable amino and fatty acids, which might be furtherly reused by host cells [42]. However, some viruses also present the ability to make use of the fatty/amino acids released from the damaged cells for the purposes of their own replication [43][44][45]. A selective autophagy of damaged cells is partly regulated by the nerve growth factor (NGF)/ tropomyosin receptor kinase A (TrkA) axis with NGF playing a cytoprotective role [31][46][47][48]. The RSV causes alterations in the expression of the NGF and its receptors, upregulating the NGF and TrkA and downregulating the p75(NTR), and thus protecting against virus-induced apoptosis; the mechanism was reported in distal but not in the proximal airway epithelium. When endogenous NGF diminished, bronchial epithelial cell survival was decreased [49]. Chakraborty reported an enhanced RSV infectivity in human bronchial epithelial cells exposed to titanium-dioxide nanoparticles (TiO2-NP) [31]. A preexposure to the TiO2-NP prior to an RSV infection upregulates the NGF/TrkA axis, inducing autophagy, which promotes cell necrosis and, as a result, a viral replication [31]. A co-exposure to the RSV and TiO2-NP results in an increased necrosis at the expense of a reduced apoptosis [31]. To the contrary, an increased apoptosis and, in consequence, a lowered viral load was reached with an experimental use of wortmannin, which is a pharmacological inhibitor of the early autophagosomal gene beclin-1 [31].
Similarly to nanoparticles, a preexposure to cigarette smoke extract also resulted in a higher viral load in human tracheobronchial epithelial (hTBE) cells due to a smaller degree of apoptosis [37]. In the study by Groskreutz, ELISA and TUNEL detection of apoptosis were used and revealed a decreased apoptosis as a result of inhibited caspases activation following an RSV infection [37]. Although the apoptosis in the airway epithelium was decreased, some of the cells were dying, probably due to necrosis, which promotes viral replication [37][50]. A decrease in apoptosis, on the other hand, can be reduced by pretreatment with N-acetylcysteine and aldehyde dehydrogenase, a fact that strongly suggests it is primarily mediated by reactive aldehydes, similar to the aforementioned reactive oxygen species (ROS) or acrolein [37].
2.2.2. Decreased Antiviral Defense
Epithelium
Activation of the epithelium mediated by interferons (IFN) is one of the baseline antiviral defense mechanisms. IFN type I (IFN α and β) and IFN type III (IFN-Γ) act through different receptors, but its physiological activity overlaps and executes antiviral responses [51]. IFN-γ is produced mostly by T cells and NK (natural killer) cells and binds to the receptor, causing an activation of JAK-STAT (Janus kinase-signal transducer and activator of transcription) cascade; as a result, phosphorylated and dimerized Stat1 translocates to the nucleus, where it binds to the specific IFN-γ activated sequence in various genes that are being induced in order to promote antiviral activity (recruitment of immune cells, antigen presentation, cell proliferation, or apoptosis) [40][52][53]. A lack of or decreased immune response might result in enhanced viral infection. While a pretreatment with IFN-γ prior to an RSV infection resulted in a decreased viral gene mRNA expression in primary human tracheobronchial epithelial cells under the influence of cigarette smoke extract (CSE), the decrease, however, was inhibited by CSE; the same effect was observed for RSV proteins expression [40]. CSE inhibits the aforementioned IFN-γ-dependent gene expression in epithelial cells via inhibition of the signal transducer and activator of transcription 1 (Stat1) phosphorylation [40]. Of interest, antiviral effects of IFN-γ blocked by CSE might be restored by gluthathione supplementation with the N-acetylcysteine or glutathione monoethyl ester (GSH-MEE) [40].
Similarly, reduced levels of IFN-γ, alongside reduced IL-12, were observed in side-stream cigarette smoke (SS)-exposed mice challenged (twice) with RSV, and in accordance with the previous study, it also led to a higher RSV gene expression [41]. Unexpectedly, Phaybouth et al. noted a similar level of decrease in Clara cell secretory protein levels in the lungs of both SS-exposed and air-exposed mice after RSV reinfection; the authors speculate that a primary infection in the neonatal period may influence an immune response in the case of a reinfection [41].
Non-ciliated airway epithelial (Clara) cells in humans are mostly located in the bronchiolar epithelium and present the ability to secret the Clara cell secretory protein (CCSP, or CC-10, or CC-16) [36][54]. CCSP is an abundant immunomodulatory protein and its deficiency results in increased viral persistence, lung inflammation, airway reactivity, and mucus production elicited by an RSV infection (the results can be reversed by restoring CCSP [36][55]. Under the influence of diesel engine emissions (DEE), the number of Clara cells (which produce CCSP) was diminished, CCSP production was attenuated, and the decrease was more accentuated after a high-level of DEE exposure [36]. Clara cells also produce other immune defense cells, for example, SP-A (surfactant protein A, a member of the collectin family), which acts as an opsonin for bacteria and viruses, and DEE exposure decreased SP-A staining in epithelial and alveolar type II cells in a dose-independent manner [36]. As a result, a significantly higher RSV gene expression was observed in mice exposed to diesel engine emissions (DEE), and the exact influence of various pollutants needs to be established, since DEE contained a number of pollutants (PM, NOx, CO, and SO2) [36].
Alveolar Macrophages (AM)
Alveolar macrophages play a crucial role in the clearance of alveolar space; AM clear the airways from both infectious agents (bacteria and viruses) and air pollutants, especially particulate matter [56]. Human AM, but also AM in mice or guinea pigs are permissive to an RSV infection, and its permissiveness is inversely related to the maturation of AM, i.e., the more mature the AM are, the lower the degree of the RSV application [57][58][59]. The role of the alveolar macrophages (AM) in RSV uptake was demonstrated in guinea pig alveolar macrophages, where RSV yield, defined as the amount of viral replication/RSV-immunopositive cell, was decreased in AM exposed to PM10 [39]. A less effective elimination of the RSV due to a decreased viral uptake by AM (a decrease reaching up to 50%) was reported by Becker and Soukup in a study investigating the human bronchial epithelial cell line and human AM from bronchoalveolar lavage, BAL (obtained from volunteers); the study also reported changes in chemokine/cytokine secretion [60]. Upon an RSV infection, AM secrete IL-8, MIP-1α, MIP-1β, and MCP-1, while RANTES derives solely from the RSV-infected bronchial epithelial cells (and is decreased in the presence of AM) [60]. PM10 alone stimulates the release of granulocyte chemoattractant IL-8, and MIP-1α, but not MIP-1β, MCP-1 or RANTES [60]. A co-exposure to PM10 and RSV inhibits MCP-1 production, and does not exhibit an additive effect on MIP-1α and IL-8 levels, which can be interpreted as a decreased production of these chemokines as well, and does not affect the RANTES production (except for the decrease in the presence of AM) [60]. The role of the chemokines must be interpreted with caution here; while MCP-1 plays a significant role in monocyte chemotaxis and differentiation of T lymphocytes, its increased expression might be related with Th2-dependent airway hyperreactivity [61]. A strong emphasis needs to be put here on the differences between the diminished immune response resulting in an attenuated virus elimination by the macrophages, and an excessive immune response leading to airway hyperreactivity, since in part, they are mediated by common mechanisms.
Dendritic Cells
Plasmacytoid dendritic cells (pDC) play an important role in antiviral protection; a viral stimulation of the Toll-like receptor (TLR) agonists initiate a secretion of type I IFN by pDC [62][63][64]. The Type I interferon system integrates early antiviral and immunostimulatory activities [65]. RSV products (viral RNA or virus intermediate RNA; vRNA or iRNA, respectively) may enter the endosomes of pDC (which contain TLR) via the process of autophagy; then, viral nucleic acids activate TLR7, thus recruiting MyD88, which causes phosphorylation of IRF7 [65]. IRF7 that is not phosphorylated and cannot be translocated into the nucleus and cannot initiate the transcription of the type I IFN genes. Cigarette smoke prevents the TLR7 activation by viral nucleic acids, it may also alter the activation of TLR7 upon trafficking into late endosomes/lysosomes; in turn, the nuclear factor (NF)- kB is activated, thus preventing the production of inflammatory cytokines (IL-1β, for example) [65]. Cigarette smoke inhibits TLR-7 and -9 expression and stimulation by specific TLR agonists in pDC [65]. Finally, cigarette smoke decreases IFN-α production in pDC in response to RSV [65]. In addition, an RSV-induced release of IL-1β, IL-10, and CXCL10 is decreased, without changes in other cytokines and chemokines (like IL-6, TNF-α, CCL2, MIP-1α= CCL3=, RANTES= CCL5 and CXCL8) [65]. A study by Castro shows a severe suppression of crucial pDC functions after cigarette smoke exposure, with an inhibition of the production of IFN-α, IL-10, IL1β, and CXCL10 and a downregulation of TLR7 [65]. Of note, another study focusing on the PM10 exposure found no statistically significant influence on the secretion of IL-1β by human airway epithelial cells challenged with RSV [66].
In another series of experiments, an influence of nitric oxide on human monocyte-derived dendritic cells (MoDCs) was shown [38]. MoDCs were exposed to the RSV during the epidemic season; then, the cultures were maintained and fresh MoDCs added monthly, and outside of the RSV epidemic season, they were exposed to nitric oxide (NO), NO donors, and NO inhibitors [38]. The exposure to any of the above agents resulted in an induction of the RSV replication, showing that the virus may remain dormant and be activated by exogenous NO [38]. The authors postulate that NO might be responsible for RSV seasonality; when higher NO levels are present in the environment, an RSV replication is triggered, similarly, patients exposed to cigarette smoke, for example, may experience an RSV infection outside the season [38]. Endogenously produced NO is expected to show antiviral activity decreasing the RSV replication, and a decreased release of nitric oxide (NO) from human monocytes (deriving from blood donors) was driven mainly by cigarette smoke; however, a higher percentage of extreme NO decrease was noticed in the presence of an RSV + CSE co-exposure, compared to each stimulus alone [67][68][69][70]. Exogenous NO (contained in cigarette smoke, for example) might decrease intracellular NO production and facilitate an infection [38]. Of note, persistently infected MoDCs exhibited an increased cell survival, suggesting that the RSV persistence involves the inhibition of apoptosis [38].
- (B)
-
Viral load fluctuations
The exposure to air pollutants may also result in lowered virus copies, but it may have a temporary character [71][72]. A lower viral load was observed in cigarette smoke- exposed compared to filtered air-exposed mice on day 4 post RSV infection; however, it was much higher on day 14. The authors discuss the possibility of type I IFN response induced by cigarette smoke which initially suppresses the replication, but due to a waning response, a delayed clearance might be expected [72].
Similarly, RSV titers were lower on days 2–4 in mice exposed to carbon black in the research by Lambert [71], but by day 7, an exacerbation of infection was observed, including an increased expression of the mRNA of proinflammatory cytokines, and protein levels of TNF-α and IL-13 in the lungs [71]. Initially, the tumor necrosis factor-α (TNF-α) levels were decreased on days 1–2, together with decreased IFN-γ mRNA, IFN-γ-inducible protein (IP-10), and lymphotactin [71].
While the IFN-γ and TNF-α show an antiviral activity, IP-10 deriving from macrophages mediates a Th1 immunity, and activates and chemoattracts neutrophils which interact with CXCR3 (IP-10R) on Th1 cells [73][74]. During an acute phase of an RSV infection, the number of CXCR3-positive cells is significantly decreased, while its ligand levels, IP-10, are elevated [75]. After preexposure to carbon black, a decrease in IP-10 was observed, suggesting that a preference towards a Th2 allergic immune response in place of a Th1 antimicrobial defense might be expected and facilitate a further inflammation in response to the RSV infection [71].
- (C)
-
Virus release
A potential reason for a worsening viral infection is an enhanced virus spread; nevertheless, researchers found no proof on the facilitated release of infectious RSV molecules. An experiment on human alveolar macrophages (AM) exposed to ozone and infected with the RSV showed no differences in the amount of infectious RSV released on day 2 and 4, neither was a percentage of the infected AM altered by the pollutant [76]. Nitrogen dioxide study revealed no differences in the release of the infectious virus on day 2 by lower doses of NO2, and, intriguingly, it was decreased in cells exposed to higher doses of NO2 [77].
2.3. An Inappropriate Host Reaction
While a diminished immune response may result in a facilitated viral replication, impaired or delayed virus elimination, an exuberant inflammatory response provokes airway hyperresponsiveness (AHR), prolonged lung infiltration and/or tissue remodelling.
2.3.1. Inflammation
In the aforementioned in vitro model by Cruz-Sanchez and colleagues, an increased secretion of IL-6 and IL-8 was observed when the RSV was harboured by PM10 (see Figure 2) [35]. In vivo studies also reveal an exacerbated inflammatory response; a murine model of RSV pneumonia disclosed an augmented inflammation in animals that were previously exposed to TiO2-NP [78]. The levels of IFN-γ and RANTES (CCL5) were used as markers of the pneumonia severity in the investigation, and their values were increased in bronchoalveolar lavage fluids (BALF) from the mice, alongside with increased IL-10, whereas viral titers were not affected [78].
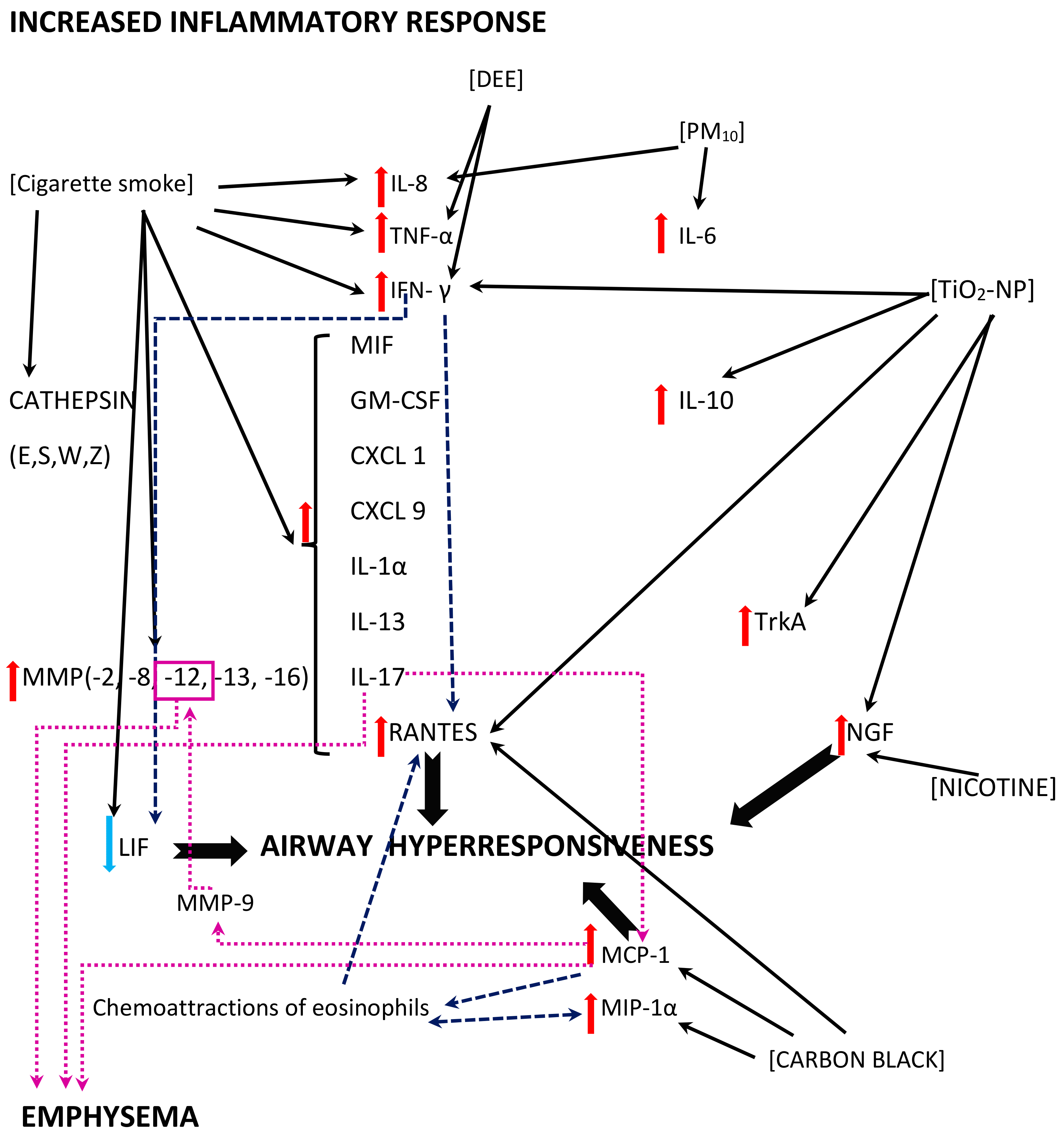
Figure 2. A diagram of the exuberant inflammatory response and its effects; multidirectional interactions between the air pollutants (shown in square brackets) and inflammatory mediators, including cytokines and chemokines are shown. The black solid line arrows reflect direct effects of the air pollutants revealed in the studies; the dashed navy blue line arrows reflect additional effects of the inflammatory mediators or immune cells contributing to the effects; the dotted magenta line arrows reflect the mechanisms related to the pathomechanism of emphysema.
Please note: the figure has only a demonstrative character and is based on the literature found for the purposes of this entry. The effects of the air pollutants might vary with regards to the presence/absence of the mechanism, its extent, and its effects. The mechanisms might be influenced by other, not verified or not shown pathways. The mechanisms might be dose-dependent, time-dependent, exposition sequence-dependent, and model-dependent (i.e., differences between the models used in the studies and human beings might be seen). The figure is simplified, and, for example, cigarette smoke is a common name for possibly different exposures (the studies used, inter alia, cigarette smoke extract, cigarette smoke condensate, or non-specified cigarette smoke exposure for different periods of time); for details, see the text and/or refer to the original articles. In addition, contradictory effects of the same air pollutant might be reported, depending on the study model.
Abbreviations: DEE—diesel engine emissions, PM10—particulate matter smaller than or equal to 10 µm in diameter, TiO2-NP—titanium-dioxide nanoparticles; CXCL (1,9)—chemokine (C-X-C motif) ligand (1, 9), GM-CSF—Granulocyte-macrophage colony-stimulating factor, IL- (1α, 6, 8, 10, 13, 17)—Interleukin- (1α, 6, 8, 10, 13, 17), IFN-γ—interferon γ, LIF—leukemia inhibitory factor, MCP-1—monocyte chemoattractant protein-1 (=CCL2), MIP-1α—macrophage inflammatory protein-1 α (=CCL3), MIF—macrophage migration inhibitory factor, MMP-(2, 8, 9, 12, 13, 16)—matrix metalloproteinase-(2, 8, 9, 12, 13, 16), NGF—nerve growth factor, RANTES—regulated upon activation, normal T-cell expressed and secreted (=CCL5), TNFα—tumor necrosis factor α, and TrkA—tropomyosin receptor kinase A.
IFN-γ and TNF-α concentrations were increased after DEE exposure, and higher DEE doses tended to influence the IFN-γ levels to a higher extent than that of TNF-α [36]. This findings are in contrast to the previously described decreased TNF-α and IFN-γ levels found after carbon black exposure; however, as stated before, an equilibrium between a beneficial immune response and exuberant response is crucial [71]. An increased release of TNF-α from human monocytes with a strong additive effect was observed after cigarette smoke extract exposure and RSV infection, and a higher percentage of extreme TNF-α increase was seen in the presence of an RSV + CSE co-exposure, compared to single exposures [67].
The vast effects of cigarette smoke exposure were observed in a murine model of repeated RSV infections [79]. A co-exposure provoked an increased in the expression of cytokines (IL-1a, IL-17, IFN-c, KC, IL-13, CXCL9, RANTES, MIF, and GM-CSF), as well as proteases (MMP-2, -8, -12, -13, -16, and cathepsins E, S, W, and Z) [79]. Protein phosphatase 2A (PP2A) and protein tyrosine phosphate 1B (PTP1B) seem to play a significant part in both effects, as they neutralize inflammation and protease expression [79]. This finding was further explored in a study on Ptp1b-deficient mice showing damage in epithelial cell barriers, an enhanced influx of immune cells and cytokine production, and increased apoptosis [80].
Interestingly, except for the pathogen-associated molecular pattern (PAMP) triggered by RSV, the damage-associated molecular pattern (DAMP) also seems to be involved in the mechanism [80][81][82]. DAMP consists of an inflammatory response induced by molecules released from infected, damaged, or dead cells; in this entry, an increased expression of S100A9 was shown in the lungs of Ptp1b -/- mice [80][83]. Under normal conditions, PTP1B suppresses S100A9 expression during an RSV infection, while an enhanced secretion of S100A9 with resulting inflammation was seen in wild-type mice exposed to cigarette smoke as a result of a desensitized PTP1B activity, as well as in differentiated human bronchial epithelial cells from COPD donors after an RSV infection [80]. S100A9 induces a cytokine release (MCP-1, CXCL10, IL-8, RANTES, G-CSF) from small airway epithelial cells in a TLR4 -dependent manner, enhancing lung damage [80]. In fact, Ptp1b-deficient mice showed an increased RSV-induced immune cell influx, damaged epithelial cell barriers, and an increased apoptosis [80]. The use of anti-S100A9 antibody reduced the immune cell influx into the lungs and perivascular inflammation, as well as apoptosis [80].
IL-8 gene and protein expression was also increased in airway epithelial cells after co-exposure to cigarette smoke concentrate (CSC) and RSV infection, together with the augmented monocyte chemoattractant protein-1 (MCP-1) expression [84]. The interferon stimulatory response element (ISRE) site of the IL-8 promoter plays a crucial role in the mechanism; an activation of the interferon regulatory factor-1 and 7 (IRF-1 and IRF-7), which bind to this region, is enhanced in response to a CSC + RSV co-stimulation, thus promoting IL-8 gene transcription [84]. The nuclear factor kappa B (NF-kB) also binds to the IL-8 promoter, synergistically augmenting the IL-8 gene transcription upon a CSC + RSV co-exposure [84]. A CSC enhanced NF-kB–driven IL-8 gene transcription is observed not only after 6 h (like in the case of RSV alone), but also 12 and 24 h post infection [84]. Therefore, an exuberant immune response may be stimulated by two different pathways, and may persist longer than in the case of a single stimuli [84].
References
- Available online: https://www.who.int/news-room/questions-and-answers/item/who-global-air-quality-guidelines (accessed on 24 August 2022).
- Lelieveld, J.; Evans, J.S.; Fnais, M.; Giannadaki, D.; Pozzer, A. The Contribution of Outdoor Air Pollution Sources to Premature Mortality on a Global Scale. Nature 2015, 525, 367–371.
- Bernstein, J.A.; Alexis, N.; Barnes, C.; Bernstein, I.L.; Nel, A.; Peden, D.; Diaz-Sanchez, D.; Tarlo, S.M.; Williams, B. Health Effects of Air Pollution. J. Allergy Clin. Immunol. 2004, 114, 1116–1123.
- Bourdrel, T.; Bind, M.-A.; Béjot, Y.; Morel, O.; Argacha, J.-F. Cardiovascular Effects of Air Pollution. Arch. Cardiovasc. Dis. 2017, 110, 634–642.
- IARC. Available online: https://monographs.iarc.who.int/list-of-classifications (accessed on 24 August 2022).
- Li, Y.; Wang, X.; Blau, D.M.; Caballero, M.T.; Feikin, D.R.; Gill, C.J.; Madhi, S.A.; Omer, S.B.; Simões, E.A.F.; Campbell, H.; et al. Global, Regional, and National Disease Burden Estimates of Acute Lower Respiratory Infections Due to Respiratory Syncytial Virus in Children Younger than 5 Years in 2019: A Systematic Analysis. Lancet 2022, 399, 2047–2064.
- Simões, E.A.F. Respiratory Syncytial Virus Disease in Young Children and Older Adults in Europe: A Burden and Economic Perspective. J. Infect. Dis. 2022, 226 (Suppl. S1), S1–S9.
- Ali, A.; Lopardo, G.; Scarpellini, B.; Stein, R.T.; Ribeiro, D. Systematic Review on Respiratory Syncytial Virus Epidemiology in Adults and the Elderly in Latin America. Int. J. Infect. Dis. 2020, 90, 170–180.
- Boattini, M.; Almeida, A.; Christaki, E.; Marques, T.M.; Tosatto, V.; Bianco, G.; Iannaccone, M.; Tsiolakkis, G.; Karagiannis, C.; Maikanti, P.; et al. Severity of RSV Infection in Southern European Elderly Patients during Two Consecutive Winter Seasons (2017–2018). J. Med. Virol. 2021, 93, 5152–5157.
- Korsten, K.; Adriaenssens, N.; Coenen, S.; Butler, C.C.; Verheij, T.J.M.; Bont, L.J.; Wildenbeest, J.G.; Butler, C.; Nair, H.; Campbell, H.; et al. World Health Organization Influenza-Like Illness Underestimates the Burden of Respiratory Syncytial Virus Infection in Community-Dwelling Older Adults. J. Infect. Dis. 2022, 226 (Suppl. S1), S71–S78.
- Mao, Z.; Li, X.; Korsten, K.; Bont, L.; Butler, C.; Wildenbeest, J.; Coenen, S.; Hens, N.; Bilcke, J.; Beutels, P.; et al. Economic Burden and Health-Related Quality of Life of Respiratory Syncytial Virus and Influenza Infection in European Community-Dwelling Older Adults. J. Infect. Dis. 2022, 226 (Suppl. S1), S87–S94.
- Bowser, D.M.; Rowlands, K.R.; Hariharan, D.; Gervasio, R.M.; Buckley, L.; Halasa-Rappel, Y.; Glaser, E.L.; Nelson, C.B.; Shepard, D.S. Cost of Respiratory Syncytial Virus Infections in US Infants: Systematic Literature Review and Analysis. J. Infect. Dis. 2022, 226 (Suppl. S2), S225–S235.
- Suh, M.; Movva, N.; Jiang, X.; Reichert, H.; Bylsma, L.C.; Fryzek, J.P.; Nelson, C.B. Respiratory Syncytial Virus Burden and Healthcare Utilization in United States Infants <1 Year of Age: Study of Nationally Representative Databases, 2011–2019. J. Infect. Dis. 2022, 226, S184–S194.
- Mazur, N.I.; Terstappen, J.; Baral, R.; Bardají, A.; Beutels, P.; Buchholz, U.J.; Cohen, C.; Crowe, J.E.; Cutland, C.L.; Eckert, L.; et al. Respiratory Syncytial Virus Prevention within Reach: The Vaccine and Monoclonal Antibody Landscape. Lancet Infect. Dis. 2022. Epub ahead of print.
- Vandini, S.; Corvaglia, L.; Alessandroni, R.; Aquilano, G.; Marsico, C.; Spinelli, M.; Lanari, M.; Faldella, G. Respiratory Syncytial Virus Infection in Infants and Correlation with Meteorological Factors and Air Pollutants. Ital. J. Pediatr. 2013, 39, 1.
- Carugno, M.; Dentali, F.; Mathieu, G.; Fontanella, A.; Mariani, J.; Bordini, L.; Milani, G.P.; Consonni, D.; Bonzini, M.; Bollati, V.; et al. PM10 Exposure Is Associated with Increased Hospitalizations for Respiratory Syncytial Virus Bronchiolitis among Infants in Lombardy, Italy. Environ. Res. 2018, 166, 452–457.
- Horne, B.D.; Joy, E.A.; Hofmann, M.G.; Gesteland, P.H.; Cannon, J.B.; Lefler, J.S.; Blagev, D.P.; Korgenski, E.K.; Torosyan, N.; Hansen, G.I.; et al. Short-Term Elevation of Fine Particulate Matter Air Pollution and Acute Lower Respiratory Infection. Am. J. Respir. Crit. Care Med. 2018, 198, 759–766.
- Karr, C.J.; Rudra, C.B.; Miller, K.A.; Gould, T.R.; Larson, T.; Sathyanarayana, S.; Koenig, J.Q. Infant Exposure to Fine Particulate Matter and Traffic and Risk of Hospitalization for RSV Bronchiolitis in a Region with Lower Ambient Air Pollution. Environ. Res. 2009, 109, 321–327.
- Nenna, R.; Evangelisti, M.; Frassanito, A.; Scagnolari, C.; Pierangeli, A.; Antonelli, G.; Nicolai, A.; Arima, S.; Moretti, C.; Papoff, P.; et al. Respiratory Syncytial Virus Bronchiolitis, Weather Conditions and Air Pollution in an Italian Urban Area: An Observational Study. Environ. Res. 2017, 158, 188–193.
- Ye, Q.; Fu, J.-F.; Mao, J.-H.; Shang, S.-Q. Haze Is a Risk Factor Contributing to the Rapid Spread of Respiratory Syncytial Virus in Children. Environ. Sci. Pollut. Res. 2016, 23, 20178–20185.
- Wrotek, A.; Badyda, A.; Czechowski, P.; Owczarek, T.; Dąbrowiecki, P.; Jackowska, T. Air Pollutants’ Concentrations Are Associated with Increased Number of RSV Hospitalizations in Polish Children. J. Clin. Med. 2021, 10, 3224.
- Radhakrishnan, D.; Ouedraogo, A.; Shariff, S.Z.; McNally, J.D.; Benchimol, E.I.; Clemens, K.K. The Association between Climate, Geography and Respiratory Syncitial Virus Hospitalizations among Children in Ontario, Canada: A Population-Based Study. BMC Infect. Dis. 2020, 20, 157.
- Lanari, M.; Giovannini, M.; Giuffré, L.; Marini, A.; Rondini, G.; Rossi, G.A.; Merolla, R.; Zuccotti, G.V.; Salvioli, G.P. Prevalence of Respiratory Syncytial Virus Infection in Italian Infants Hospitalized for Acute Lower Respiratory Tract Infections, and Association between Respiratory Syncytial Virus Infection Risk Factors and Disease Severity. Pediatr. Pulmonol. 2002, 33, 458–465.
- Nguyen, S.N.; Nguyen, T.N.T.; Vu, L.T.; Nguyen, T.D. Clinical Epidemiological Characteristics and Risk Factors for Severe Bronchiolitis Caused by Respiratory Syncytial Virus in Vietnamese Children. Int. J. Pediatr. 2021, 2021, 9704666.
- Maedel, C.; Kainz, K.; Frischer, T.; Reinweber, M.; Zacharasiewicz, A. Increased Severity of Respiratory Syncytial Virus Airway Infection Due to Passive Smoke Exposure. Pediatr. Pulmonol. 2018, 53, 1299–1306.
- Thomson, N.C. The Role of Smoking in Asthma and Chronic Obstructive Pulmonary Disease Overlap. Immunol. Allergy Clin. N. Am. 2022, 42, 615–630.
- Sikkel, M.B.; Quint, J.K.; Mallia, P.; Wedzicha, J.A.; Johnston, S.L. Respiratory Syncytial Virus Persistence in Chronic Obstructive Pulmonary Disease. Pediatr. Infect. Dis. J. 2008, 27 (Suppl. S10), S63–S70.
- Loaiza-Ceballos, M.C.; Marin-Palma, D.; Zapata, W.; Hernandez, J.C. Viral Respiratory Infections and Air Pollutants. Air Qual. Atmos. Health 2022, 15, 105–114.
- Smallcombe, C.C.; Harford, T.J.; Linfield, D.T.; Lechuga, S.; Bokun, V.; Piedimonte, G.; Rezaee, F. Titanium Dioxide Nanoparticles Exaggerate Respiratory Syncytial Virus-Induced Airway Epithelial Barrier Dysfunction. Am. J. Physiol. Cell. Mol. Physiol. 2020, 319, L481–L496.
- Rezaee, F.; Harford, T.J.; Linfield, D.T.; Altawallbeh, G.; Midura, R.J.; Ivanov, A.I.; Piedimonte, G. cAMP-Dependent Activation of Protein Kinase a Attenuates Respiratory Syncytial Virus-Induced Human Airway Epithelial Barrier Disruption. PLoS ONE 2017, 12, e0181876.
- Chakraborty, S.; Castranova, V.; Perez, M.K.; Piedimonte, G. Nanoparticles Increase Human Bronchial Epithelial Cell Susceptibility to Respiratory Syncytial Virus Infection via Nerve Growth Factor-Induced Autophagy. Physiol. Rep. 2017, 5, e13344.
- Ambalavanan, N.; Stanishevsky, A.; Bulger, A.; Halloran, B.; Steele, C.; Vohra, Y.; Matalon, S. Titanium Oxide Nanoparticle Instillation Induces Inflammation and Inhibits Lung Development in Mice. Am. J. Physiol. Cell. Mol. Physiol. 2013, 304, L152–L161.
- Kobayashi, N.; Naya, M.; Endoh, S.; Maru, J.; Yamamoto, K.; Nakanishi, J. Comparative Pulmonary Toxicity Study of Nano-TiO(2) Particles of Different Sizes and Agglomerations in Rats: Different Short- and Long-Term Post-instillation Results. Toxicology 2009, 264, 110–118.
- Morimoto, Y.; Kobayashi, N.; Shinohara, N.; Myojo, T.; Tanaka, I.; Nakanishi, J. Hazard Assessments of Manufactured Nanomaterials. J. Occup. Health 2010, 52, 325–334.
- Cruz-Sanchez, T.M.; Haddrell, A.E.; Hackett, T.L.; Singhera, G.K.; Marchant, D.; Lekivetz, R.; Meredith, A.; Horne, D.; Knight, D.A.; van Eeden, S.F.; et al. Formation of a Stable Mimic of Ambient Particulate Matter Containing Viable Infectious Respiratory Syncytial Virus and Its Dry-Deposition Directly onto Cell Cultures. Anal. Chem. 2013, 85, 898–906.
- Harrod, K.S.; Jaramillo, R.J.; Rosenberger, C.L.; Wang, S.-Z.; Berger, J.A.; McDonald, J.D.; Reed, M.D. Increased Susceptibility to RSV Infection by Exposure to Inhaled Diesel Engine Emissions. Am. J. Respir. Cell Mol. Biol. 2003, 28, 451–463.
- Groskreutz, D.J.; Monick, M.M.; Babor, E.C.; Nyunoya, T.; Varga, S.M.; Look, D.C.; Hunninghake, G.W. Cigarette Smoke Alters Respiratory Syncytial Virus–Induced Apoptosis and Replication. Am. J. Respir. Cell Mol. Biol. 2009, 41, 189–198.
- Hobson, L.; Everard, M.L. Persistent of Respiratory Syncytial Virus in Human Dendritic Cells and Influence of Nitric Oxide. Clin. Exp. Immunol. 2008, 151, 359–366.
- Kaan, P.M.; Hegele, R.G. Interaction between Respiratory Syncytial Virus and Particulate Matter in Guinea Pig Alveolar Macrophages. Am. J. Respir. Cell Mol. Biol. 2003, 28, 697–704.
- Modestou, M.A.; Manzel, L.J.; El-Mahdy, S.; Look, D.C. Inhibition of IFN-γ-Dependent Antiviral Airway Epithelial Defense by Cigarette Smoke. Respir. Res. 2010, 11, 64.
- Phaybouth, V.; Wang, S.-Z.; Hutt, J.A.; McDonald, J.D.; Harrod, K.S.; Barrett, E.G. Cigarette Smoke Suppresses Th1 Cytokine Production and Increases RSV Expression in a Neonatal Model. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2006, 290, L222–L231.
- Morris, S.; Swanson, M.; Lieberman, A.; Reed, M.; Yue, Z.; Lindell, D.M.; Lukacs, N.W. Autophagy-Mediated Dendritic Cell Activation Is Essential for Innate Cytokine Production and APC Function with Respiratory Syncytial Virus Responses. J. Immunol. 2011, 187, 3953–3961.
- Nakashima, A.; Tanaka, N.; Tamai, K.; Kyuuma, M.; Ishikawa, Y.; Sato, H.; Yoshimori, T.; Saito, S.; Sugamura, K. Survival of Parvovirus B19-Infected Cells by Cellular Autophagy. Virology 2006, 349, 254–263.
- Huang, S.-C.; Chang, C.-L.; Wang, P.-S.; Tsai, Y.; Liu, H.-S. Enterovirus 71-Induced Autophagy Detected In Vitro and In Vivo Promotes Viral Replication. J. Med. Virol. 2009, 81, 1241–1252.
- Heaton, N.S.; Randall, G. Dengue Virus and Autophagy. Viruses 2011, 3, 1332–1341.
- Rosso, P.; Moreno, S.; Fracassi, A.; Rocco, M.L.; Aloe, L. Nerve Growth Factor and Autophagy: Effect of Nasal Anti-NGF-Antibodies Administration on Ambra1 and Beclin-1 Expression in Rat Brain. Growth Factors 2015, 33, 401–409.
- Franco, M.L.; Melero, C.; Sarasola, E.; Acebo, P.; Luque, A.; Calatayud-Baselga, I.; García-Barcina, M.; Vilar, M. Mutations in TrkA Causing Congenital Insensitivity to Pain with Anhidrosis (CIPA) Induce Misfolding, Aggregation, and Mutation-dependent Neurodegeneration by Dysfunction of the Autophagic Flux. J. Biol. Chem. 2016, 291, 21363–21374.
- Wang, Z.-G.; Li, H.; Huang, Y.; Li, R.; Wang, X.-F.; Yu, L.-X.; Guang, X.-Q.; Li, L.; Zhang, H.-Y.; Zhao, Y.-Z.; et al. Nerve Growth Factor-Induced Akt/mTOR Activation Protects the Ischemic Heart via Restoring Autophagic Flux and Attenuating Ubiquitinated Protein Accumulation. Oncotarget 2017, 8, 5400–5413.
- Othumpangat, S.; Gibson, L.; Samsell, L.; Piedimonte, G. NGF Is an Essential Survival Factor for Bronchial Epithelial Cells during Respiratory Syncytial Virus Infection. PLoS ONE 2009, 4, e6444.
- Cuff, S.; Ruby, J. Evasion of Apoptosis by DNA Viruses. Immunol. Cell Biol. 1996, 74, 527–537.
- Lazear, H.M.; Schoggins, J.W.; Diamond, M.S. Shared and Distinct Functions of Type I and Type III Interferons. Immunity 2019, 50, 907–923.
- Bach, E.A.; Aguet, M.; Schreiber, R.D. The IFN γ Receptor: A Paradigm for Cytokine Receptor Signaling. Annu. Rev. Immunol. 1997, 15, 563–591.
- Greenlund, A.C.; Morales, M.O.; Viviano, B.L.; Yan, H.; Krolewski, J.; Schreiber, R.D. Stat Recruitment by Tyrosine-Phosphorylated Cytokine Receptors: An Ordered Reversible Affinity-Driven Process. Immunity 1995, 2, 677–687.
- Widdicombe, J.G.; Pack, R.J. The Clara Cell. Eur. J. Respir. Dis. 1982, 63, 202–220.
- Wang, S.-Z.; Rosenberger, C.L.; Bao, Y.-X.; Stark, J.M.; Harrod, K. Clara Cell Secretory Protein Modulates Lung Inflammatory and Immune Responses to Respiratory Syncytial Virus Infection. J. Immunol. 2003, 171, 1051–1060.
- Miyata, R.; van Eeden, S.F. The Innate and Adaptive Immune Response Induced by Alveolar Macrophages Exposed to Ambient Particulate Matter. Toxicol. Appl. Pharmacol. 2011, 257, 209–226.
- Panuska, J.R.; Cirino, N.M.; Midulla, F.; Despot, J.E.; McFadden, E.R.; Huang, Y.T. Productive Infection of Isolated Human Alveolar Macrophages by Respiratory Syncytial Virus. J. Clin. Investig. 1990, 86, 113–119.
- Franke, G.; Freihorst, J.; Steinmüller, C.; Verhagen, W.; Hockertz, S.; Lohmann-Matthes, M.-L. Interaction of Alveolar Macrophages and Respiratory Syncytial Virus. J. Immunol. Methods 1994, 174, 173–184.
- Dakhama, A.; Kaan, P.M.; Hegele, R.G. Permissiveness of Guinea Pig Alveolar Macrophage Subpopulations to Acute Respiratory Syncytial Virus Infection In Vitro. Chest 1998, 114, 1681–1688.
- Becker, S.; Soukup, J.M. Exposure to Urban Air Particulates Alters the Macrophage-Mediated Inflammatory Response to Respiratory Viral Infection. J. Toxicol. Environ. Health A 1999, 57, 445–457.
- Giuffrida, M.J.; Valero, N.; Mosquera, J.; De Mon, M.A.; Chacín, B.; Espina, L.M.; Gotera, J.; Bermudez, J.; Mavarez, A. Increased Cytokine/Chemokines in Serum from Asthmatic and Non-asthmatic Patients with Viral Respiratory Infection. Influ. Other Respir. Viruses 2014, 8, 116–122.
- Cella, M.; Jarrossay, D.; Facchetti, F.; Alebardi, O.; Nakajima, H.; Lanzavecchia, A.; Colonna, M. Plasmacytoid Monocytes Migrate to Inflamed Lymph Nodes and Produce Large Amounts of Type I Interferon. Nat. Med. 1999, 5, 919–923.
- Fitzgerald-Bocarsly, P.; Dai, J.; Singh, S. Plasmacytoid Dendritic Cells and Type I IFN: 50 Years of Convergent History. Cytokine Growth Factor Rev. 2008, 19, 3–19.
- Siegal, F.P.; Kadowaki, N.; Shodell, M.; Fitzgerald-Bocarsly, P.A.; Shah, K.; Ho, S.; Antonenko, S.; Liu, Y.-J. The Nature of the Principal Type 1 Interferon-Producing Cells in Human Blood. Science 1999, 284, 1835–1837.
- Castro, S.M.; Chakraborty, K.; Guerrero-Plata, A. Cigarette Smoke Suppresses TLR-7 Stimulation in Response to Virus Infection in Plasmacytoid Dendritic Cells. Toxicol. Vitr. 2011, 25, 1106–1113.
- Hirota, J.A.; Marchant, D.J.; Singhera, G.K.; Moheimani, F.; Dorscheid, D.R.; Carlsten, C.; Sin, D.; Knight, D. Urban Particulate Matter Increases Human Airway Epithelial Cell IL-1β Secretion Following Scratch Wounding and H1N1 Influenza A Exposure In Vitro. Exp. Lung Res. 2015, 41, 353–362.
- Raza, M.W.; Essery, S.D.; Weir, D.M.; Ogilvie, M.M.; Elton, R.A.; Blackwell, C.C. Infection with Respiratory Syncytial Virus and Water-Soluble Components of Cigarette Smoke Alter Production of Tumour Necrosis Factor α and Nitric Oxide by Human Blood Monocytes. FEMS Immunol. Med. Microbiol. 1999, 24, 387–394.
- Domachowske, J.B.; Ali-Ahmad, D.; Bonville, C.A.; Rosenberg, H.F. Replication of Respiratory Syncytial Virus Is Inhibited in Target Cells Generating Nitric Oxide In Situ. Front. Biosci. 2003, 8, a48–a53.
- Stark, J.M.; Khan, A.M.; Chiappetta, C.L.; Xue, H.; Alcorn, J.L.; Colasurdo, G.N. Immune and Functional Role of Nitric Oxide in a Mouse Model of Respiratory Syncytial Virus Infection. J. Infect. Dis. 2005, 191, 387–395.
- Colasurdo, G.N.; Fullmer, J.J.; Elidemir, O.; Atkins, C.; Khan, A.M.; Stark, J.M. Respiratory Syncytial Virus Infection in a Murine Model of Cystic Fibrosis. J. Med. Virol. 2006, 78, 651–658.
- Lambert, A.L.; Trasti, F.S.; Mangum, J.B.; Everitt, J.I. Effect of Preexposure to Ultrafine Carbon Black on Respiratory Syncytial Virus Infection in Mice. Toxicol. Sci. 2003, 72, 331–338.
- Mebratu, Y.A.; Smith, K.R.; Agga, G.E.; Tesfaigzi, Y. Inflammation and Emphysema in Cigarette Smoke-Exposed Mice When Instilled with Poly (I:C) or Infected with Influenza A or Respiratory Syncytial Viruses. Respir. Res. 2016, 17, 75.
- Mestan, J.; Digel, W.; Mittnacht, S.; Hillen, H.; Blohm, D.; Möller, A.; Jacobsen, H.; Kirchner, H. Antiviral Effects of Recombinant Tumour Necrosis Factor In Vitro. Nature 1986, 323, 816–819.
- Neville, L.F.; Mathiak, G.; Bagasra, O. The Immunobiology of Interferon-γ Inducible Protein 10 kD (IP-10): A Novel, Pleiotropic Member of the C-X-C Chemokine Superfamily. Cytokine Growth Factor Rev. 1997, 8, 207–219.
- Roe, M.F.E.; Bloxham, D.M.; Cowburn, A.S.; O’Donnell, D.R. Changes in Helper Lymphocyte Chemokine Receptor Expression and Elevation of IP-10 During Acute Respiratory Syncytial Virus Infection in Infants. Pediatr. Allergy Immunol. 2011, 22, 229–234.
- Soukup, J.; Koren, H.; Becker, S. Ozone Effect on Respiratory Syncytial Virus Infectivity and Cytokine Production by Human Alveolar Macrophages. Environ. Res. 1993, 60, 178–186.
- Becker, S.; Soukup, J.M. Effect of Nitrogen Dioxide on Respiratory Viral Infection in Airway Epithelial Cells. Environ. Res. 1999, 81, 159–166.
- Hashiguchi, S.; Yoshida, H.; Akashi, T.; Komemoto, K.; Ueda, T.; Ikarashi, Y.; Miyauchi, A.; Konno, K.; Yamanaka, S.; Hirose, A.; et al. Titanium Dioxide Nanoparticles Exacerbate Pneumonia in Respiratory Syncytial Virus (RSV)-Infected Mice. Environ. Toxicol. Pharmacol. 2015, 39, 879–886.
- Foronjy, R.F.; Dabo, A.J.; Taggart, C.C.; Weldon, S.; Geraghty, P. Respiratory Syncytial Virus Infections Enhance Cigarette Smoke Induced COPD in Mice. PLoS ONE 2014, 9, e90567.
- Foronjy, R.F.; Ochieng, P.O.; Salathe, M.; Dabo, A.J.; Eden, E.; Baumlin, N.; Cummins, N.; Barik, S.; Campos, M.; Thorp, E.; et al. Protein Tyrosine Phosphatase 1B Negatively Regulates S100A9-Mediated Lung Damage during Respiratory Syncytial Virus Exacerbations. Mucosal Immunol. 2016, 9, 1317–1329.
- Xu, H.; An, H.; Hou, J.; Han, C.; Wang, P.; Yu, Y.; Cao, X. Phosphatase PTP1B Negatively Regulates MyD88- and TRIF-Dependent Proinflammatory Cytokine and Type I Interferon Production in TLR-Triggered Macrophages. Mol. Immunol. 2008, 45, 3545–3552.
- Medgyesi, D.; Hobeika, E.; Biesen, R.; Kollert, F.; Taddeo, A.; Voll, R.E.; Hiepe, F.; Reth, M. The Protein Tyrosine Phosphatase PTP1B is a Negative Regulator of CD40 and BAFF-R Signaling and Controls B Cell Autoimmunity. J. Exp. Med. 2014, 211, 427–440.
- Tsai, S.-Y.; Segovia, J.A.; Chang, T.-H.; Morris, I.R.; Berton, M.T.; Tessier, P.A.; Tardif, M.R.; Cesaro, A.; Bose, S. DAMP Molecule S100A9 Acts as a Molecular Pattern to Enhance Inflammation during Influenza A Virus Infection: Role of DDX21-TRIF-TLR4-MyD88 Pathway. PLoS Pathog. 2014, 10, e1003848.
- Castro, S.M.; Kolli, D.; Guerrero-Plata, A.; Garofalo, R.P.; Casola, A. Cigarette Smoke Condensate Enhances Respiratory Syncytial Virus–Induced Chemokine Release by Modulating NF-kappa B and Interferon Regulatory Factor Activation. Toxicol. Sci. 2008, 106, 509–518.
More
Information
Subjects:
Respiratory System
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.1K
Revisions:
2 times
(View History)
Update Date:
01 Nov 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No