Respiratory syncytial virus (RSV) is one of the major infectious agents in paediatrics, and its relationship with air pollution is frequently observed. A relationship between lower air quality and RSV infections was observed mainly in hospital-based and some community-based studies, with particular interest in PM2.5,However, the molecular basis of this interaction is sparsely reported. We sought to systematically review the existing body PM10,of nlitric dioxide (NO2), ozone,erature carbon monoxide (CO), and benzene. Exposurend identify the knowledge gaps to another pollutant, cigarette smoke, is a well-described risk factor for a more severe swer the question: which molecular mechanisms are implied in the air pollutants–RSV infection in children, an increased RSV LRTI prevalence, and chronic obstructive pulmonary disease (COPD); in the latter case, the RSV can be persistently present, escaping an immune responseteraction? Online databases were searched for original studies published before August 2022 focusing on molecular mechanisms of the interaction.
- respiratory syncytial virus
- air pollution
- particulate matter
- PM2.5
- pathomechanism
- environmental pollutants
- bronchial hyperreactivity
1. Introduction
2. Molecular Mechanisms
2.1. A Facilitated Viral Entry
2.1.1. Epithelial Barrier
Epithelium forms a first-line barrier in the airways that is crucial for antiviral defense, and a damage to the airway epithelium is characteristic for an RSV infection [29][30][52,61]. The RSV has been previously shown to cause a disintegration of the epithelium and its increased permeability (so called “leaky epithelium”), mainly due to a disruption of the apical junctional complexes (AJC), which are intercellular complexes [30][61]. Except for a diminished integrity, the RSV also causes a remodeling of the actin cytoskeletal to which AJC is linked [29][30][52,61]. The RSV-induced epithelial disintegration might be enhanced by an exposure to nanoparticles and titanium-dioxide nanoparticles (TiO2-NP), which was shown in a number of studies to be a suitable model for the studies on pulmonary effects of environmental nanoparticles (see Figure 15) [29][31][32][33][34][36,52,62,63,64]. Both in vitro (immortalized human bronchial epithelial cells) and in vivo (murine) models of TiO2-NP effects, investigated by Smallcombe, confirmed the hypothesis of an augmented barrier dysfunction [29][52]. The barrier dysfunction comprised an amplified AJC disruption and led to an increased viral replication [29][52]. The molecular mechanism of the epithelium integrity disruption by TiO2-NP was shown to be related to an oxidative stress and a generation of reactive oxygen species (ROS) and a pretreatment with an antioxidant (N-acetylcysteine) not only attenuated the AJC disruption, but was also able to reverse the enhanced RSV infection [29][52].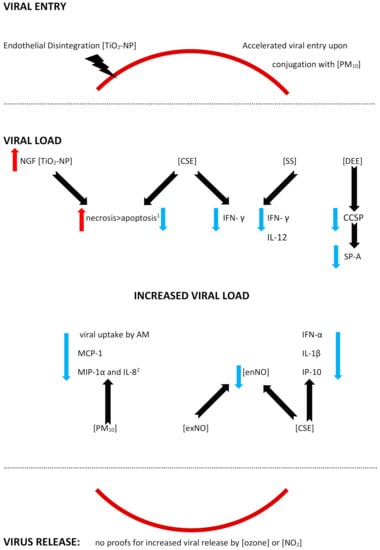
2.1.2. A Facilitated Viral Entry
A facilitated viral entry was reported in an in vitro mimic of PM10 containing the RSV that were deposited onto the airway epithelial cells [35][37]. As a result, an accelerated viral entry was observed upon the RSV conjugation to PM, and the mechanism involved an endocytic pathway [35][37]. Moreover, the RSV survival was increased when it was associated with PM10 in the model [35][37]. No studies proving enhanced viral receptors expression on the respiratory tract cells were found.2.2. An Altered Viral Load
- (A)
-
An Increased Viral Load
2.2.1. Autophagy, Decreased Apoptosis, and Enhanced Necrosis
Autophagy is crucial for the immune response, since the structures of damaged cells might be cut into recyclable amino and fatty acids, which might be furtherly reused by host cells [42][65]. However, some viruses also present the ability to make use of the fatty/amino acids released from the damaged cells for the purposes of their own replication [43][44][45][66,67,68]. A selective autophagy of damaged cells is partly regulated by the nerve growth factor (NGF)/ tropomyosin receptor kinase A (TrkA) axis with NGF playing a cytoprotective role [31][46][47][48][36,69,70,71]. The RSV causes alterations in the expression of the NGF and its receptors, upregulating the NGF and TrkA and downregulating the p75(NTR), and thus protecting against virus-induced apoptosis; the mechanism was reported in distal but not in the proximal airway epithelium. When endogenous NGF diminished, bronchial epithelial cell survival was decreased [49][72]. Chakraborty reported an enhanced RSV infectivity in human bronchial epithelial cells exposed to titanium-dioxide nanoparticles (TiO2-NP) [31][36]. A preexposure to the TiO2-NP prior to an RSV infection upregulates the NGF/TrkA axis, inducing autophagy, which promotes cell necrosis and, as a result, a viral replication [31][36]. A co-exposure to the RSV and TiO2-NP results in an increased necrosis at the expense of a reduced apoptosis [31][36]. To the contrary, an increased apoptosis and, in consequence, a lowered viral load was reached with an experimental use of wortmannin, which is a pharmacological inhibitor of the early autophagosomal gene beclin-1 [31][36]. Similarly to nanoparticles, a preexposure to cigarette smoke extract also resulted in a higher viral load in human tracheobronchial epithelial (hTBE) cells due to a smaller degree of apoptosis [37][40]. In the study by Groskreutz, ELISA and TUNEL detection of apoptosis were used and revealed a decreased apoptosis as a result of inhibited caspases activation following an RSV infection [37][40]. Although the apoptosis in the airway epithelium was decreased, some of the cells were dying, probably due to necrosis, which promotes viral replication [37][50][40,73]. A decrease in apoptosis, on the other hand, can be reduced by pretreatment with N-acetylcysteine and aldehyde dehydrogenase, a fact that strongly suggests it is primarily mediated by reactive aldehydes, similar to the aforementioned reactive oxygen species (ROS) or acrolein [37][40].2.2.2. Decreased Antiviral Defense
Epithelium
Activation of the epithelium mediated by interferons (IFN) is one of the baseline antiviral defense mechanisms. IFN type I (IFN α and β) and IFN type III (IFN-Γ) act through different receptors, but its physiological activity overlaps and executes antiviral responses [51][74]. IFN-γ is produced mostly by T cells and NK (natural killer) cells and binds to the receptor, causing an activation of JAK-STAT (Janus kinase-signal transducer and activator of transcription) cascade; as a result, phosphorylated and dimerized Stat1 translocates to the nucleus, where it binds to the specific IFN-γ activated sequence in various genes that are being induced in order to promote antiviral activity (recruitment of immune cells, antigen presentation, cell proliferation, or apoptosis) [40][52][53][49,75,76]. A lack of or decreased immune response might result in enhanced viral infection. While a pretreatment with IFN-γ prior to an RSV infection resulted in a decreased viral gene mRNA expression in primary human tracheobronchial epithelial cells under the influence of cigarette smoke extract (CSE), the decrease, however, was inhibited by CSE; the same effect was observed for RSV proteins expression [40][49]. CSE inhibits the aforementioned IFN-γ-dependent gene expression in epithelial cells via inhibition of the signal transducer and activator of transcription 1 (Stat1) phosphorylation [40][49]. Of interest, antiviral effects of IFN-γ blocked by CSE might be restored by gluthathione supplementation with the N-acetylcysteine or glutathione monoethyl ester (GSH-MEE) [40][49]. Similarly, reduced levels of IFN-γ, alongside reduced IL-12, were observed in side-stream cigarette smoke (SS)-exposed mice challenged (twice) with RSV, and in accordance with the previous study, it also led to a higher RSV gene expression [41][50]. Unexpectedly, Phaybouth et al. noted a similar level of decrease in Clara cell secretory protein levels in the lungs of both SS-exposed and air-exposed mice after RSV reinfection; the authors speculate that a primary infection in the neonatal period may influence an immune response in the case of a reinfection [41][50]. Non-ciliated airway epithelial (Clara) cells in humans are mostly located in the bronchiolar epithelium and present the ability to secret the Clara cell secretory protein (CCSP, or CC-10, or CC-16) [36][54][29,77]. CCSP is an abundant immunomodulatory protein and its deficiency results in increased viral persistence, lung inflammation, airway reactivity, and mucus production elicited by an RSV infection (the results can be reversed by restoring CCSP [36][55][29,78]. Under the influence of diesel engine emissions (DEE), the number of Clara cells (which produce CCSP) was diminished, CCSP production was attenuated, and the decrease was more accentuated after a high-level of DEE exposure [36][29]. Clara cells also produce other immune defense cells, for example, SP-A (surfactant protein A, a member of the collectin family), which acts as an opsonin for bacteria and viruses, and DEE exposure decreased SP-A staining in epithelial and alveolar type II cells in a dose-independent manner [36][29]. As a result, a significantly higher RSV gene expression was observed in mice exposed to diesel engine emissions (DEE), and the exact influence of various pollutants needs to be established, since DEE contained a number of pollutants (PM, NOx, CO, and SO2) [36][29].Alveolar Macrophages (AM)
Alveolar macrophages play a crucial role in the clearance of alveolar space; AM clear the airways from both infectious agents (bacteria and viruses) and air pollutants, especially particulate matter [56][79]. Human AM, but also AM in mice or guinea pigs are permissive to an RSV infection, and its permissiveness is inversely related to the maturation of AM, i.e., the more mature the AM are, the lower the degree of the RSV application [57][58][59][80,81,82]. The role of the alveolar macrophages (AM) in RSV uptake was demonstrated in guinea pig alveolar macrophages, where RSV yield, defined as the amount of viral replication/RSV-immunopositive cell, was decreased in AM exposed to PM10 [39][45]. A less effective elimination of the RSV due to a decreased viral uptake by AM (a decrease reaching up to 50%) was reported by Becker and Soukup in a study investigating the human bronchial epithelial cell line and human AM from bronchoalveolar lavage, BAL (obtained from volunteers); the study also reported changes in chemokine/cytokine secretion [60][33]. Upon an RSV infection, AM secrete IL-8, MIP-1α, MIP-1β, and MCP-1, while RANTES derives solely from the RSV-infected bronchial epithelial cells (and is decreased in the presence of AM) [60][33]. PM10 alone stimulates the release of granulocyte chemoattractant IL-8, and MIP-1α, but not MIP-1β, MCP-1 or RANTES [60][33]. A co-exposure to PM10 and RSV inhibits MCP-1 production, and does not exhibit an additive effect on MIP-1α and IL-8 levels, which can be interpreted as a decreased production of these chemokines as well, and does not affect the RANTES production (except for the decrease in the presence of AM) [60][33]. The role of the chemokines must be interpreted with caution here; while MCP-1 plays a significant role in monocyte chemotaxis and differentiation of T lymphocytes, its increased expression might be related with Th2-dependent airway hyperreactivity [61][83]. A strong emphasis needs to be put here on the differences between the diminished immune response resulting in an attenuated virus elimination by the macrophages, and an excessive immune response leading to airway hyperreactivity, since in part, they are mediated by common mechanisms.Dendritic Cells
Plasmacytoid dendritic cells (pDC) play an important role in antiviral protection; a viral stimulation of the Toll-like receptor (TLR) agonists initiate a secretion of type I IFN by pDC [62][63][64][84,85,86]. The Type I interferon system integrates early antiviral and immunostimulatory activities [65][35]. RSV products (viral RNA or virus intermediate RNA; vRNA or iRNA, respectively) may enter the endosomes of pDC (which contain TLR) via the process of autophagy; then, viral nucleic acids activate TLR7, thus recruiting MyD88, which causes phosphorylation of IRF7 [65][35]. IRF7 that is not phosphorylated and cannot be translocated into the nucleus and cannot initiate the transcription of the type I IFN genes. Cigarette smoke prevents the TLR7 activation by viral nucleic acids, it may also alter the activation of TLR7 upon trafficking into late endosomes/lysosomes; in turn, the nuclear factor (NF)- kB is activated, thus preventing the production of inflammatory cytokines (IL-1β, for example) [65][35]. Cigarette smoke inhibits TLR-7 and -9 expression and stimulation by specific TLR agonists in pDC [65][35]. Finally, cigarette smoke decreases IFN-α production in pDC in response to RSV [65][35]. In addition, an RSV-induced release of IL-1β, IL-10, and CXCL10 is decreased, without changes in other cytokines and chemokines (like IL-6, TNF-α, CCL2, MIP-1α= CCL3=, RANTES= CCL5 and CXCL8) [65][35]. A study by Castro shows a severe suppression of crucial pDC functions after cigarette smoke exposure, with an inhibition of the production of IFN-α, IL-10, IL1β, and CXCL10 and a downregulation of TLR7 [65][35]. Of note, another study focusing on the PM10 exposure found no statistically significant influence on the secretion of IL-1β by human airway epithelial cells challenged with RSV [66][42]. In another series of experiments, an influence of nitric oxide on human monocyte-derived dendritic cells (MoDCs) was shown [38][43]. MoDCs were exposed to the RSV during the epidemic season; then, the cultures were maintained and fresh MoDCs added monthly, and outside of the RSV epidemic season, they were exposed to nitric oxide (NO), NO donors, and NO inhibitors [38][43]. The exposure to any of the above agents resulted in an induction of the RSV replication, showing that the virus may remain dormant and be activated by exogenous NO [38][43]. The authors postulate that NO might be responsible for RSV seasonality; when higher NO levels are present in the environment, an RSV replication is triggered, similarly, patients exposed to cigarette smoke, for example, may experience an RSV infection outside the season [38][43]. Endogenously produced NO is expected to show antiviral activity decreasing the RSV replication, and a decreased release of nitric oxide (NO) from human monocytes (deriving from blood donors) was driven mainly by cigarette smoke; however, a higher percentage of extreme NO decrease was noticed in the presence of an RSV + CSE co-exposure, compared to each stimulus alone [67][68][69][70][53,87,88,89]. Exogenous NO (contained in cigarette smoke, for example) might decrease intracellular NO production and facilitate an infection [38][43]. Of note, persistently infected MoDCs exhibited an increased cell survival, suggesting that the RSV persistence involves the inhibition of apoptosis [38][43].- (B)
-
Viral load fluctuations
- (C)
-
Virus release
2.3. An Inappropriate Host Reaction
While a diminished immune response may result in a facilitated viral replication, impaired or delayed virus elimination, an exuberant inflammatory response provokes airway hyperresponsiveness (AHR), prolonged lung infiltration and/or tissue remodelling.2.3.1. Inflammation
In the aforementioned in vitro model by Cruz-Sanchez and colleagues, an increased secretion of IL-6 and IL-8 was observed when the RSV was harboured by PM10 (see Figure 26) [35][37]. In vivo studies also reveal an exacerbated inflammatory response; a murine model of RSV pneumonia disclosed an augmented inflammation in animals that were previously exposed to TiO2-NP [78][41]. The levels of IFN-γ and RANTES (CCL5) were used as markers of the pneumonia severity in the investigation, and their values were increased in bronchoalveolar lavage fluids (BALF) from the mice, alongside with increased IL-10, whereas viral titers were not affected [78][41].Please note: the figure has only a demonstrative character and is based on the literature found for the purposes of this rentryview. The effects of the air pollutants might vary with regards to the presence/absence of the mechanism, its extent, and its effects. The mechanisms might be influenced by other, not verified or not shown pathways. The mechanisms might be dose-dependent, time-dependent, exposition sequence-dependent, and model-dependent (i.e., differences between the models used in the studies and human beings might be seen). The figure is simplified, and, for example, cigarette smoke is a common name for possibly different exposures (the studies used, inter alia, cigarette smoke extract, cigarette smoke condensate, or non-specified cigarette smoke exposure for different periods of time); for details, see the text and/or refer to the original articles. In addition, contradictory effects of the same air pollutant might be reported, depending on the study model. Abbreviations: DEE—diesel engine emissions, PM10—particulate matter smaller than or equal to 10 µm in diameter, TiO2-NP—titanium-dioxide nanoparticles; CXCL (1,9)—chemokine (C-X-C motif) ligand (1, 9), GM-CSF—Granulocyte-macrophage colony-stimulating factor, IL- (1α, 6, 8, 10, 13, 17)—Interleukin- (1α, 6, 8, 10, 13, 17), IFN-γ—interferon γ, LIF—leukemia inhibitory factor, MCP-1—monocyte chemoattractant protein-1 (=CCL2), MIP-1α—macrophage inflammatory protein-1 α (=CCL3), MIF—macrophage migration inhibitory factor, MMP-(2, 8, 9, 12, 13, 16)—matrix metalloproteinase-(2, 8, 9, 12, 13, 16), NGF—nerve growth factor, RANTES—regulated upon activation, normal T-cell expressed and secreted (=CCL5), TNFα—tumor necrosis factor α, and TrkA—tropomyosin receptor kinase A. IFN-γ and TNF-α concentrations were increased after DEE exposure, and higher DEE doses tended to influence the IFN-γ levels to a higher extent than that of TNF-α [36][29]. This findings are in contrast to the previously described decreased TNF-α and IFN-γ levels found after carbon black exposure; however, as stated before, an equilibrium between a beneficial immune response and exuberant response is crucial [71][46]. An increased release of TNF-α from human monocytes with a strong additive effect was observed after cigarette smoke extract exposure and RSV infection, and a higher percentage of extreme TNF-α increase was seen in the presence of an RSV + CSE co-exposure, compared to single exposures [67][53]. The vast effects of cigarette smoke exposure were observed in a murine model of repeated RSV infections [79][38]. A co-exposure provoked an increased in the expression of cytokines (IL-1a, IL-17, IFN-c, KC, IL-13, CXCL9, RANTES, MIF, and GM-CSF), as well as proteases (MMP-2, -8, -12, -13, -16, and cathepsins E, S, W, and Z) [79][38]. Protein phosphatase 2A (PP2A) and protein tyrosine phosphate 1B (PTP1B) seem to play a significant part in both effects, as they neutralize inflammation and protease expression [79][38]. This finding was further explored in a study on Ptp1b-deficient mice showing damage in epithelial cell barriers, an enhanced influx of immune cells and cytokine production, and increased apoptosis [80][39]. Interestingly, except for the pathogen-associated molecular pattern (PAMP) triggered by RSV, the damage-associated molecular pattern (DAMP) also seems to be involved in the mechanism [80][81][82][39,93,94]. DAMP consists of an inflammatory response induced by molecules released from infected, damaged, or dead cells; in this enstrudy, an increased expression of S100A9 was shown in the lungs of Ptp1b -/- mice [80][83][39,95]. Under normal conditions, PTP1B suppresses S100A9 expression during an RSV infection, while an enhanced secretion of S100A9 with resulting inflammation was seen in wild-type mice exposed to cigarette smoke as a result of a desensitized PTP1B activity, as well as in differentiated human bronchial epithelial cells from COPD donors after an RSV infection [80][39]. S100A9 induces a cytokine release (MCP-1, CXCL10, IL-8, RANTES, G-CSF) from small airway epithelial cells in a TLR4 -dependent manner, enhancing lung damage [80][39]. In fact, Ptp1b-deficient mice showed an increased RSV-induced immune cell influx, damaged epithelial cell barriers, and an increased apoptosis [80][39]. The use of anti-S100A9 antibody reduced the immune cell influx into the lungs and perivascular inflammation, as well as apoptosis [80][39]. IL-8 gene and protein expression was also increased in airway epithelial cells after co-exposure to cigarette smoke concentrate (CSC) and RSV infection, together with the augmented monocyte chemoattractant protein-1 (MCP-1) expression [84][34]. The interferon stimulatory response element (ISRE) site of the IL-8 promoter plays a crucial role in the mechanism; an activation of the interferon regulatory factor-1 and 7 (IRF-1 and IRF-7), which bind to this region, is enhanced in response to a CSC + RSV co-stimulation, thus promoting IL-8 gene transcription [84][34]. The nuclear factor kappa B (NF-kB) also binds to the IL-8 promoter, synergistically augmenting the IL-8 gene transcription upon a CSC + RSV co-exposure [84][34]. A CSC enhanced NF-kB–driven IL-8 gene transcription is observed not only after 6 h (like in the case of RSV alone), but also 12 and 24 h post infection [84][34]. Therefore, an exuberant immune response may be stimulated by two different pathways, and may persist longer than in the case of a single stimuli [84][34].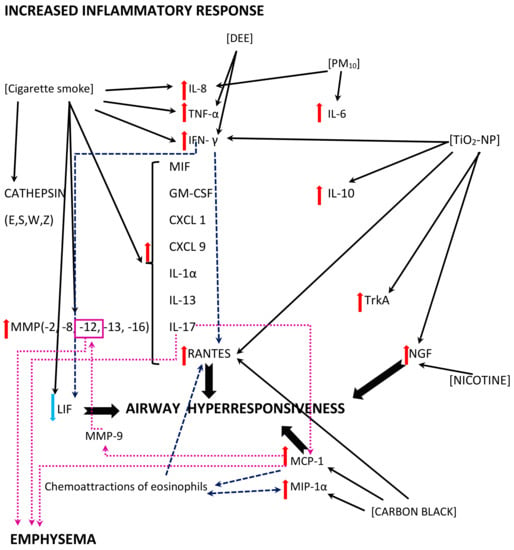 Figure 26. A diagram of the exuberant inflammatory response and its effects; multidirectional interactions between the air pollutants (shown in square brackets) and inflammatory mediators, including cytokines and chemokines are shown. The black solid line arrows reflect direct effects of the air pollutants revealed in the studies; the dashed navy blue line arrows reflect additional effects of the inflammatory mediators or immune cells contributing to the effects; the dotted magenta line arrows reflect the mechanisms related to the pathomechanism of emphysema.
Figure 26. A diagram of the exuberant inflammatory response and its effects; multidirectional interactions between the air pollutants (shown in square brackets) and inflammatory mediators, including cytokines and chemokines are shown. The black solid line arrows reflect direct effects of the air pollutants revealed in the studies; the dashed navy blue line arrows reflect additional effects of the inflammatory mediators or immune cells contributing to the effects; the dotted magenta line arrows reflect the mechanisms related to the pathomechanism of emphysema.