Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ginevra Micangeli | -- | 4938 | 2022-10-28 15:08:25 | | | |
| 2 | Dean Liu | -1 word(s) | 4937 | 2022-10-31 02:46:31 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Micangeli, G.; Menghi, M.; Profeta, G.; Tarani, F.; Mariani, A.; Petrella, C.; Barbato, C.; Ferraguti, G.; Ceccanti, M.; Tarani, L.; et al. Impact of Oxidative Stress on Pediatrics Syndromes. Encyclopedia. Available online: https://encyclopedia.pub/entry/31840 (accessed on 27 July 2026).
Micangeli G, Menghi M, Profeta G, Tarani F, Mariani A, Petrella C, et al. Impact of Oxidative Stress on Pediatrics Syndromes. Encyclopedia. Available at: https://encyclopedia.pub/entry/31840. Accessed July 27, 2026.
Micangeli, Ginevra, Michela Menghi, Giovanni Profeta, Francesca Tarani, Alessandro Mariani, Carla Petrella, Christian Barbato, Giampiero Ferraguti, Mauro Ceccanti, Luigi Tarani, et al. "Impact of Oxidative Stress on Pediatrics Syndromes" Encyclopedia, https://encyclopedia.pub/entry/31840 (accessed July 27, 2026).
Micangeli, G., Menghi, M., Profeta, G., Tarani, F., Mariani, A., Petrella, C., Barbato, C., Ferraguti, G., Ceccanti, M., Tarani, L., & Fiore, M. (2022, October 28). Impact of Oxidative Stress on Pediatrics Syndromes. In Encyclopedia. https://encyclopedia.pub/entry/31840
Micangeli, Ginevra, et al. "Impact of Oxidative Stress on Pediatrics Syndromes." Encyclopedia. Web. 28 October, 2022.
Copy Citation
Oxidative stress is a condition determined by an imbalance between antioxidant and oxidative factors. Oxidative stress can have serious consequences on our organism. Indeed, it causes both necrosis and cell apoptosis, determining cellular aging, increased carcinogenesis, vascular stiffening, increased autoimmune diseases, and muscle decay.
oxidative stress
antioxidant
pediatrics
FASD
1. Introduction
Oxidative stress is a condition in which the balance between antioxidant and proxying factors is altered in favor of the production of oxidizing species; this determines the overproduction of radical oxygen species, defined as ROS, and can be associated with a reduction in the synthesis of antioxidant factors, as shown in Figure 1 [1][2]. In the correct balance of the cellular oxidative state, ROS are necessary for the performance of some functions [3]. In fact, among the functions performed by ROS, there are the degradation of pathogens, the regulation of cardiac and vascular activities, the regulation of intracellular calcium concentration, and the phosphorylation or the dephosphorylation of proteins [3][4]. In the context of pediatric syndromes, the role of oxidative stress is not yet fully elucidated, and there is no review of literature that compares the various types of oxidative stress in different syndromes [5].
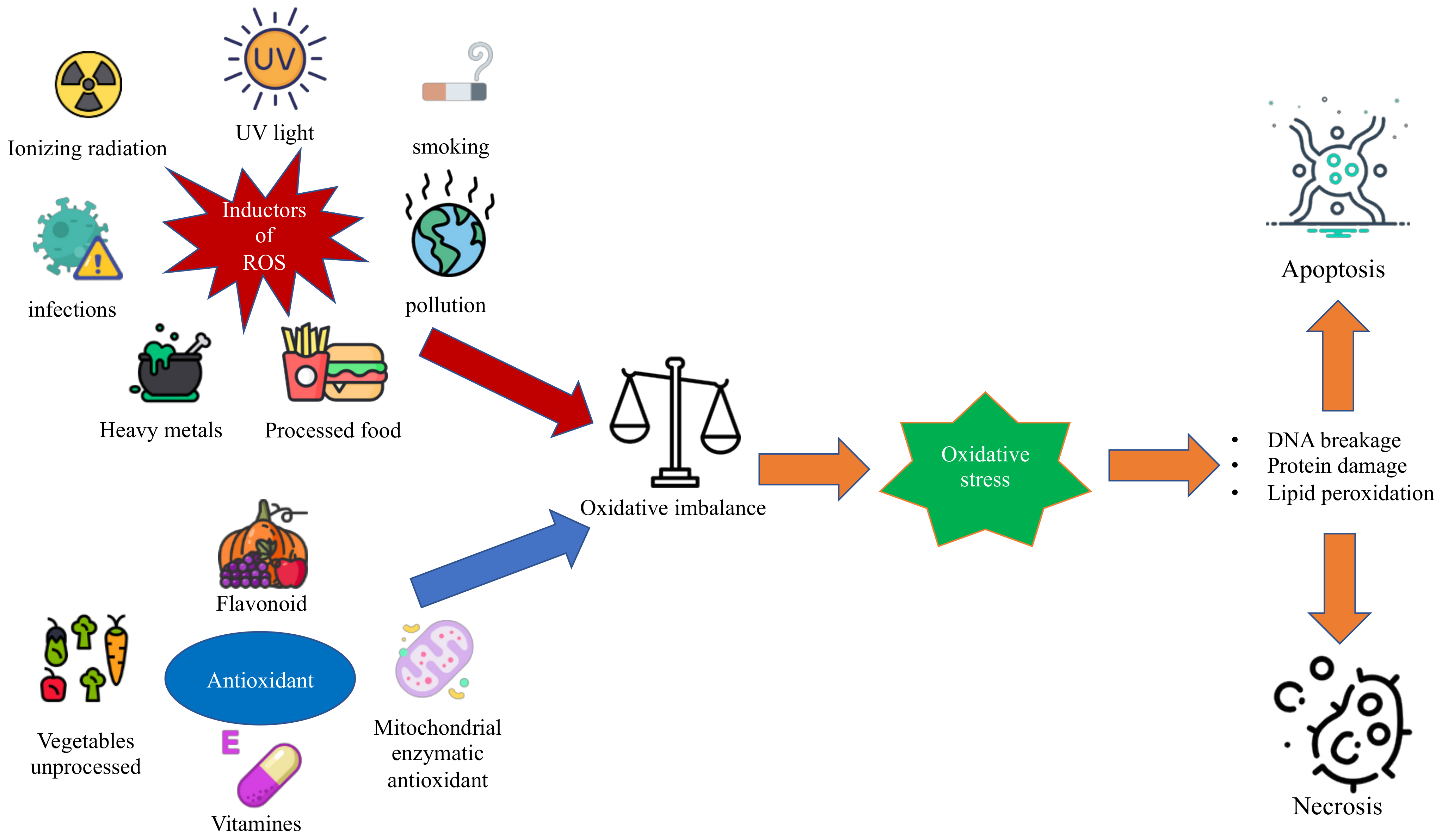
Figure 1. The figure shows the oxidative imbalance between ROS and antioxidant factors that leads to oxidative stress and the related consequences on the cell.
2. Pediatrics Syndromes Associated with Oxidative Stress
2.1. Fetal Alcohol Spectrum Disorders
FASD is the term adopted to identify the group of symptoms affecting cognitive and behavioral functioning that can be found in newborns whose mothers consumed alcohol during pregnancy [6][7][8]. It may be interesting to stress that, even if the latest Diagnostic and Statistical Manual of Mental Disorders (DSM-V) does not consider FASD to be a specific clinical mental health disorder, it does acknowledge the need for further studies on neurobehavioral disorders associated with prenatal alcohol exposure [9][10].
FASD is a disease with no genetic origin but is due solely and exclusively to the mother’s consumption of alcohol during pregnancy [11]. At present, a minimum dose determining the disease has not yet been identified. For this reason, it is absolutely forbidden to consume alcohol during pregnancy, even in minimal quantities [12].
The effect of alcohol consumption can vary extremely [13], ranging from mild intellectual and/or behavioral impairments to fetal alcohol syndrome (FAS), whose prevention has become the main goal of public health workers since the identification of the damages alcohol consumption can provoke to the fetus [14]. For instance, it should be noted that FAS has been classified as the leading cause of mental retardation worldwide and that, among neurobehavioral and developmental abnormalities, FAS would benefit the most from prevention—that is to say, the foremost preventable cause [10][15][16][17][18][19][20][21].
An interesting aspect of the matter is comparing the causal link between oxidation and FASD as opposed to other genetic disorders. In fact, the patients with FASD are conceived as perfectly healthy, and the oxidation provoked by alcohol is the cause of the pathology since it produces its effect on an otherwise physiological substrate [10][22][23].
More specifically, the scientific community has noticed a strong effect of alcohol exposure on the hippocampal proteome, culminating with the alternation of more than 600 hippocampal proteins playing important roles in the axonal growth regulation, such as annexin A2, nucleobindin-1, and glypican-4, regulators of cellular growth and developmental morphogenesis and, in the cerebellum, cadherin-13, reticulocalbin-2, and ankyrin-2 [24].
The ethanol contained in alcoholic beverages is first converted into acetaldehyde by various enzymes such as alcohol dehydrogenase (ADH), cytochrome CYP2E1, and catalase [25][26]. In the brain, the two most important pathways seem to be those mediated by cytochrome CYP2E1 and catalase, which metabolize 20% and 60% of ethanol, respectively [26]. On the other hand, the most represented pathway for the liver and stomach is mediated by alcohol dehydrogenase, which metabolizes 90% of the ethanol taken [27].
The acetaldehyde produced is then converted into acetate by acetaldehyde dehydrogenase (ALDH), which in turn is converted into acetyl-coenzyme-A in the liver [28].
A major problem of alcohol intake during pregnancy is related to the fact that the fetus has limited or null abilities in the metabolization and elimination of alcohol. Indeed, the various enzymes involved gradually increase their activity during the various stages of gestation [10]. For example, cytochrome CYP2E1 increases its activity compared to an adult, from 40% in the second semester to 80% in the third semester [10]. Therefore, the fetus is likely to be able to metabolize less than half of the alcohol taken by the mother [26].
The increase in ROS in FASD also appears to be due to NOX enzymes belonging to the NADPH-dependent family of enzymes [29]. The NOXs enzymes are expressed at the level of microglia, astrocytes, and the vascular system at the cerebral level, with an important role in the appropriate brain development [30]. The isoforms most involved in ROS production are NOX2 and NOX4 [26]. In FASD patients, it would appear that early exposure to ethanol during pregnancy would increase the activity of NOX isoforms with a significant increase in ROS, cell damage, and ultimately apoptosis [26]. This pathway, in conjunction with that related to the reduced fetal detoxification activity of CYP2E1, would seem to explain the drastic increase in ROS and the consequent phenotype of FASD patients [31].
Even if the mechanisms of the alcohol-induced neuropathology in regions of high vulnerability remain to be comprehensively determined, the teratogenic effects are thought to be the ultimate result of the ethanol-induced dysregulation of a variety of intracellular pathways, which ultimately culminate in toxicity and cell death [11]. The generation of ROS as the possible result of ethanol exposure produces an imbalance in the intracellular redox state, leading to an overall increase in oxidative stress [32]. This would explain the predominant effect that alcohol has on the brain regarding neurobehavioral impairment and deficient brain growth since brain tissue is rich in fatty acids, which chemically are the perfect substrate for the ROS [12]. The most valid biochemical explication would be that the enzyme CYP2E1, whose presence in the brain overlaps with its organogenesis, oxidizes ethanol, generating a hydroxyethyl or superoxide radical, which would target polyunsaturated fatty acid side chains in brain tissue membranes [24][31].
As a consequence, fetal brain tissue results in damage during organogenesis, manifesting neurological dysfunctions after birth [12][30][31][33].
2.2. Williams-Beuren Syndrome
Williams-Beuren Syndrome (WBS) is a rare genetic disease with multisystemic involvement, affecting nearly 1 out of 7500–10,000 people. A deletion of a group of genes, situated on chromosome 7 (7q11.23), including the ELN (elastin; OMIM *130,160) gene, is the WBS cause. These genes encode elastin, whose deletion is responsible for the cardiovascular traits and accelerated aging in patients with this disease [34][35]. The common features of this syndrome are represented by the following: facial dysmorphisms, cardiovascular malformations, endocrinological alterations, and intellectual and cognitive disturbances [34]. WBS individuals often show signs of mildly accelerated aging such as cataracts, graying of hair during adolescence, high-frequency sensorineural hearing loss, senile emphysema, premature wrinkling of the skin, and a precipitous age-associated decrease in episodic memory [35][36][37]. The oxidative stress role in these patients is less known than in other genetic disorders; nevertheless, recent studies have shown a correlation between the elastin-insufficiency of WBS and cardiovascular and respiratory diseases [38][39]. For the cardiovascular system, the major impact of ROS seems to be in the hypertension predisposition, in fact as stated before the ELN is a crucial component of the vascular wall providing recoil to elastic vessels [38][39].
Arteries with decreased ELN content are less compliant and develop structural modifications that include elevated numbers of smooth muscle and elastic lamellae; consequently, people with ELN deficiency have developmentally, rather than environmentally, elicited vascular stiffness [40]. They also show anatomical differences in branching and arterial tortuosity that lead to a turbulent flow and increased hemodynamic stress on vessel walls [40]. Recently, increasing pieces of evidence demonstrate that ROS production, particularly O2•− and H2O2, through activation of vascular NADPH oxidases, has a central role in vascular mechanotransduction [41][42]. Smooth and endothelial muscle cells express different NADPH oxidases consisting of multiple oxidases and regulatory subunits [43][44]. Other lines of evidence suggest also that hemodynamic forces can either directly or indirectly activate vascular NADPH oxidase-derived ROS production [43][44]. Abundant findings suggest that hypertension might be associated with the potentiated activity of the vascular NADPH oxidases type 1 and 2 having a regulatory subunit, the defined p47phox. This molecule is encoded by the NCF1 (neutrophil cytosolic factor 1) gene located on the telomeric region of chromosome 7 [39]. The lower expression of NCF1 is related to lower blood pressure and minor ROS production, so if the deletion on chromosome 7 extends to include the NCF1 gene, the incidence of hypertension decreases [39][45].
In the respiratory system, WBS subjects with ELN haploinsufficiency may be predisposed to the early development of pulmonary emphysema, the elastin in fact, is a key component of elastic fibers within the lung [45]. Emphysema, a subtype of chronic obstructive pulmonary disease (COPD), is characterized by progressive destruction and loss of elastic fibers, but this is not the only possible mechanism [37]. In WBS, there is a mitochondrial dysfunction. In fact, in the primary fibroblasts of patients affected, decreased basal respiration and maximal respiratory capacity were found, as well as increased ROS generation and decreased ATP synthesis [38]. This mitochondrial dysfunction may be due to the loss of DNAJC30, a gene included in the WBS critical region (WBSCR) [46]. Recent studies have uncovered significant mitochondrial signatures in chronic lung diseases, perturbations of cellular homeostatic programs associated with mitochondrial dysfunction in chronic lung diseases include modulation of the cellular autophagy program, its mitochondria-specific subtype (mitophagy), and associated changes in mitochondrial dynamics and activation of cell death pathways such as apoptosis and necrosis [28][47][48][49]. In addition, mitochondrial dysfunction may have differential and cell type-specific functional consequences in different lung cell types (e.g., epithelial cells, fibroblasts, immune cells), which may differentially impact disease progression, leading to divergent outcomes such as the development of fibrosis or emphysema [50].
Mitochondrial dysfunction could also be related to impaired brain development. Removal of DNAJC30 in mice resulted in hypofunctional mitochondria, diminished morphological features of neocortical pyramidal neurons, and altered behaviors reminiscent of WBS [5]. The mitochondrial features are consistent with the observations of decreased integrity of oxidative phosphorylation supercomplexes and ATP-synthase dimers in WBS [51]. Thus, researchers identify DNAJC30 as an auxiliary component of ATP-synthase machinery and reveal mitochondrial maladies as underlying certain defects in brain development and function associated with WBS [46].
2.3. Ataxia-Telangiectasia
Ataxia-telangiectasia (A-T) is an autosomal recessive disease eliciting several pathologies in the first two decades of life, including immunodeficiency, insulin resistance, telangiectasias, cerebellar ataxia, T-lymphoid tumors, and radiosensitivity [5][52]. Among these, T-cell malignancies and cerebellar ataxia are the most incapacitating phenotypes of this disease [5]. A-T is due to mutations in the Ataxia Telangiectasia Mutated (ATM) gene [53]. The gene encodes a serine/threonine protein kinase belonging to the phosphoinositide 3-kinase (PI3K)-related protein kinase family [53]. ATM plays a main role at the beginning of cellular responses to DNA double-strand breaks [54][55]. Nevertheless, some of the phenotypic disruptions observed in A-T individuals are not easily elucidated only by changes in DNA damage response (DDR) paths [54][55]. Since the antioxidant treatment of ATM-null mice improves intrinsic defects in stem cell renewal and might contribute to the delay of their tumor onset, it has been hypothesized that augmented accumulation of intracellular ROS, associated with ATM impairments, may contribute to these diseases [56][57].
The first possible mechanism described is mitochondrial dysfunction. In fact, the ATMs role ATM in maintaining mitochondrial functionality is well shown [28][58][59]. The ATM loss in vivo produces mitochondria disruptions, causing overproduction of ROS, a marked reduction in ATP, and ultrastructural abnormalities [57]. Furthermore, the selective exclusion of impaired mitochondria, known as mitophagy, is strongly impaired, leading to dysfunctional organelles accumulation [5]. The following other pieces of evidence have been reviewed also in neuroblastoma: the depletion of ATM produces comparable mitochondrial phenotypes and mitophagy changes [59]. Studies on thymocytes isolated from mutated mice showed mitochondrial abnormalities in ATM-deficient thymic cells, with disorganized structure and swollen appearance as well as a complex I activity significantly decreased that led to an abnormal generation of ROS taken together, demonstrating the relation between ATM deficiency and mitochondrial dysfunction [5][58].
Noteworthy A-T findings on alternative ROS sources were presented by Weyemi et al., who demonstrated that NADPH oxidase 4 (NOX4) in A-T cells could be a key instrument of oxidative stress [60]. Indeed, it was shown that NOX4, which constitutively triggers ROS in a variety of tissues and cell types as well as has a subtle role in oxidative DNA damage and the consequent senescence, was quite up-regulated in A-T individuals, comparable to normal cells with an ATM protein kinase privation [61][62]. Additionally, NOX4 is correlated with higher oxidative damage and apoptosis [60][63]. Moreover, NOX4 inactivation decreased cancer incidence (lymphoma) in ATM-deficient mice compared to control mice [60][63].
Semlitsch et al. demonstrated the following other important sources of damage in A-T patients: a specific oxidation product: the ox-LDL (oxidized low-density lipoprotein) [64][65]. OxLDL is a potent proinflammatory chemoattractant for macrophages and T-lymphocytes. It is also cytotoxic for endothelial cells and stimulates them to release soluble inflammatory molecules [66]. In addition, oxLDL has turned out to be highly immunogenic and promotes changes in cell cycle protein expression and subsequent translocation and activation of transcription factors [65][67]. These events help to perpetuate a cycle of vascular inflammation and lipid/protein dysregulation within the artery wall and also may create a cellular prothrombotic state that complicates later stages of atherosclerosis [67].
OxLDL generates oxidative stress in the vascular system induced phosphorylation of ATM and downstream activation of p21 in fibroblasts and endothelial cells. ATM-deficient cells are extremely sensitive to the toxic effects of ROS, especially H2O2 and nitric oxide developing an increased DNA fragmentation [68][69].
In conclusion, oxidative stress in A-T is related to various sources such as mitochondrial dysfunction, augmented production of ROS by the up-regulation of NOX4, and increased sensibility to ox-LDL with DNA fragmentation [63][65].
2.4. Down Syndrome
Down syndrome (DS) is a congenital disorder caused by a complete or partial trisomy of Chr21 (HSA21), and it is the most common genetic cause of significant intellectual disability, with an incidence of around 1:800 births [70]. Most DS cases (95%) are caused by non-disjunction of chromosomes in meiosis I during the formation of gametes, while 3.2% are caused by translocation, and 1.8% of residual DS cases are caused by mosaicism [71]. The effects of trisomy 21 can be very different from one individual to the next, and not every DS subject shows the same phenotypic features. The main alteration is represented by intellectual disability, other common features are congenital heart disease, Alzheimer’s disease, leukemia, hypotonia, motor disorders, and various physical anomalies [72]. Although pathological mechanisms leading to DS phenotypes are still uncertain, it is evident that the presence of the third chromosome 21 is responsible for altered development during embryogenesis and organogenesis [73]. Many of its clinical features were also studied as possible consequences of oxidative stress and cellular senescence since the change in chromosome 21 affects genes playing key roles in the redox state regulation [38][74]. When associated with other redox imbalance genetic diseases, DS has been broadly investigated [75][76]. Several findings showed that changes in proteins and genes involved in ATP consumption, mitochondrial pathways, and increased ROS production may explain the wide variety of phenotypes [77].
The most important genes that are involved in the increase in oxidative stress levels found in DS individuals and in the Ts65Dn mouse model are SOD1, APP, BACH1, Et2, S100B, and CRB [78].
The first chromosome 21 gene that was characterized and identified in different DS tissues was SOD-1 (Cu/Zn superoxide dismutase 1) (OMIM *147,450) [79]. It acts as an antioxidant defense that catalyzes the dismutation of superoxide radicals (O2−) to hydrogen peroxide (H2O2), then metabolized to water by catalases (CAT) and glutathione peroxidase (GTPx) [80]. SOD-1 was shown to be approximately 50% higher than normal in a wide range of DS tissues and cells, including B and T lymphocytes, fibroblasts, and erythrocytes [81]. Furthermore, SOD-1 overexpression in the brain was not associated with a parallel CAT and GTPx elevation, determining an imbalance in the ratio of SOD-1 to CAT and GTPx levels, leading to an H2O2 accumulation and consequent damage [82]. Surprisingly, DS tissues, including the brain, show changes in the SOD-1/GTPx activity ratio [83]. Associated with CAT and GTPx, a decreased expression of peroxiredoxin 2 was also found in the DS fetal brain, which contributes to the improved susceptibility of DS neurons to undergoing oxidative damage [84]. Since SOD-1 may play an important role in the pathogenesis of Ts21, it could be used as a potential biomarker for the prenatal diagnosis of Ts21 [85].
In addition to the well-recognized role of SOD-1, alterations in the oxidative imbalance could also be caused by the over-production of beta-amyloid (Aβ), due to the triplication of APP. APP (Amyloid Beta A4 Precursor Protein) (OMIM *104,760) encodes for a precursor protein of Aβ. Its overexpression leads to Aβ deposition and increases the formation of senile plaques, a main neuropathological finding of Alzheimer’s disease [72]. DS is the most common cause of early-onset Alzheimer’s disease-dementia [86]. In patients with DS, the increase in APP expression is strongly associated with Aβ deposition in adult life and the early and increased formation of senile plaques [76]. Oxidative stress and early plaque formation in the brain are closely connected. In fact, ROS damage increases the probability of the formation of protein aggregates as it obstructs the normal processes of protein elimination. The Aβ aggregates can be targets of oxidative processes, inserted as oligomers within the cell membrane and promoting a process of lipid peroxidation (LPO) [38].
BACH1 (BTB domain and CNC homolog 1; OMIM *602,751), encoded on Hsa21, is another key element in the regulation of the antioxidant response in DS [77]. It is a transcription repressor that inhibits selected gene transcription involved in stress response, such as heme oxygenase-1 (HO-1) and NADPH. The BACH1 overexpression potentiates ROS production from the endothelial cell’s mitochondria [77]. DS mouse model studies and investigations into DS patients demonstrated that BACH1 was significantly upregulated [87]. In DS, it is probable that BACH1 protein upregulation could block the induction of antioxidant genes, therefore eliciting increased oxidative stress in the cell [81].
The analyses of two more genes, S100β and Ets-2, both located on chromosome 21, have been reported as associated with oxidative damage.
S100β (S100 calcium-binding protein, beta; OMIM *176,990) is an astroglia-derived Ca2+-binding protein actively secreted from astrocytes that modulates the activity of neurons, microglia, astrocytes, monocytes, and endothelial cells depending on its concentration [88]. S100β increased expression in astrocytes from DS and Alzheimer’s patients was shown in association with neuritic plaques [89]. The overexpression of S100β increases ROS formation and results in increasing neuritic neuronal and APP with consequently accelerated amyloid accumulation [38].
Ets-2 (ETS Protooncogene 2, Transcription Factor) (OMIM *164,740) is a transcription factor playing crucial roles in immune responses, cancer, and bone development, and it is overexpressed in Down Syndrome [73]. Overexpression is associated with increased neuronal apoptosis [38]. Ets-2 overexpression in cultured HCN leads to activation of a mitochondrial death apoptotic pathway. In DS/AD brains, upregulation of ets-2 appears closely associated with AD neurodegenerative lesions. Chronic oxidative stress in DS and AD brains may promote ets-2 expression, which may predispose to the activation of a mitochondrial death pathway [90].
In conclusion, cognitive and neurological disorders may be the consequence of the overexpression of SOD1, APP, ETS-2, S100β, and abnormal production of BACH1. Finally, the following other main alterations that can affect individuals with DS are congenital and acquired cardiovascular abnormalities: although no specific gene has been identified, it seems that increased oxidative stress and mitochondrial dysfunction are associated with the increased development of these complications [91].
2.5. Marfan Syndrome
Marfan syndrome (MFS) is an autosomal dominant disease that affects the connective tissue with variable penetrance and an estimated prevalence of one in 10,000–20,000 individuals [92]. In 90% of cases [93], MFS is caused by mutations in Fibrillin-1 (FBN1), located on chromosome 15q21.1 and containing 65 exons [94]. The cardinal features involve the ocular and skeletal systems (e.g., tall stature, arachnodactyly, and ectopia lentis) [95], but the most life-threatening manifestations are related to cardiovascular complications, including mitral valve prolapse, arrhythmias, coronary artery disease, left ventricular hypertrophy, congestive heart failure, aortic insufficiency, dilatation of the aortic root, and aortic dissection [96][97]. Considering those cardiovascular complications, early recognition and appropriate management are critical for patients with MFS. Clinical criteria and, in particular, Ghent nosology, outlined in 2010, are used in the diagnosis of MFS [98].
It is well known that the highly reactive oxygen-derived free radicals (ROS) play an important role in the genesis and progression of various cardiovascular diseases, including arrhythmias, aortic dilatation, aortic dissection, left ventricular hypertrophy, coronary arterial disease, and congestive heart failure [99]. MFS is characterized by the presence of ascending aortic aneurysms resulting from the altered assembly of extracellular matrix fibrillin-containing microfibrils and dysfunction of TGF-β signaling [100]. It has been demonstrated that patients affected by MFS show impaired contractile function and endothelial-dependent relaxation resulting from oxidative stress in the thoracic aorta [101]. Endothelial dysfunction increases the inducible nitric oxide synthase (iNOS) pathway, leading to an excess in nitric oxide (NO) production that causes tissue damage [102]. Moreover, the results of studies have suggested that ROS could be involved in smooth muscle cell phenotype switching and apoptosis as well as matrix metalloproteinase activation, resulting in extracellular matrix (ECM) remodeling [103]. Among ROS species, Jiménez-Altayó et al. identified that H2O2 directly produced by NOX4 and/or by the transmutation of O2•− by SODs could be the most relevant ROS candidate for the Marfan-associated redox stress because unlike O2•−, H2O2 is permeable to cell membranes and has a significantly longer lifespan than O2•−, showing high reactivity for cysteine residues leading to their oxidation [104]. Oxidative stress plays an important role in the formation of the ascending aortic aneurysm but also in the evolution of the aneurysm. Studies on both animals and humans studies demonstrated that augmented redox stress is correlated to the progression of the aortic aneurysm [103]. According to some studies, the progression of thoracic aortic aneurysm is the result of the markedly impaired aortic contractile function as well as decreased nitric oxide (NO)-mediated endothelial-dependent relaxation [105]. Recently, Fiorillo et al. demonstrated, for the first time, signs of oxidative stress in the plasma of patients with MFS. Moreover, they showed a significant correlation between the intensity of oxidative stress and the severity of the clinical manifestations, suggesting systemic oxidative damage [106].
2.6. Fanconi’s Anemia
Fanconi’s anemia (FA) is an inherited pathology of DNA repair, with progressive pancytopenia, bone marrow failure, variable congenital malformations, and a predisposition to hematological or solid tumors [107]. AF is transmitted with an autosomal recessive inheritance and is due to mutations in genes involved in DNA repair and genomic stability. In total, 15 genes have been identified, these 15 genes encode proteins identified as FANCA, B, C, D1, D2, E, F, G, I, J, L, M, N, O, and P. These proteins form nuclear complexes and are activated in response to DNA damage breakage [108][109][110].
The incidence is approximately 1–5 cases per 1,000,000 inhabitants, with approximately 2000 cases described worldwide [107]. The phenotypic anomalies are many, such as aplasia of the radius, skin hyperpigmentation, microphthalmia, nystagmus, and reduced vision. Cardiac, renal, and urogenital defects and short stature, deafness, and hypogonadism can occur in a lower percentage of incidence [107]. The prognosis is unfavorable due to the higher incidence of solid tumors and leukemia [111].
In patients with FA, oxidative stress is an important determinant of some clinical conditions related to the disease. In fact, it has been seen that in affected patients there would be a reduction in the activity of SOD with consequently reduced production of antioxidant species. Furthermore, there would seem to be an increase in the production of TNF-α, which would lead to an increase in the production of O2•− with consequent apoptosis and disruption of DNA [112][113]. In fact, the increase in TNF-α would seem to lead to reduced activity of some proteins of the FANC group such as FANCA and FANCG with a reduced ability to prevent DNA damage [114]. The latter protein is expressed at the mitochondrial level and its mutation would lead to the reduced activation of a mitochondrial peroxide, PRDX3, with important antioxidant activities [113]. The FANCC protein, on the other hand, would bind cytochrome-P450 2E1 (CYP2E1), which has a detoxifying activity at the cellular level [114]. The FANCD2 protein interacts with the ATM protein and appears to stabilize it by reducing the production of radical species [112].
2.7. Autism Spectrum Disorders (ASD)
Autism spectrum disorders are defined as a group of neurodevelopmental disorders that involve alterations in social, work, school, and personal functioning [115]. They usually cause difficulties in acquiring, maintaining, and applying specific skills or sets of information [116]. The current prevalence in Italy is 15 children per 1000 with a 1:4 male-to-female ratio. In the last decade, there has been an increase in diagnoses due in part to the change in diagnostic criteria [117]. The etiopathogenesis is unknown and it is therefore believed that it may be multifactorial and dependent on both environmental and genetic factors. In recent years, the correlation between environmental pollutants and autism spectrum disorders has been studied, and what has emerged is that exposure to such substances as ionizing radiation, pesticides, and heavy metals increases the risk of developing this pathology [118]. From a clinical point of view, the symptoms are variable but usually characterized by the following: persistent deficits in social communication and interaction, repetitiveness, and sectorial behavior, interests, or activities [116]. The diagnosis is clinical and is based on the evaluation of the patient and on the execution of some tests that are generally carried out from the age of two, including the Autism Diagnostic Interview-Revised (ADI-R); Childhood Autism Rating Scale (CARS) and Autism Diagnostic Observation Schedule (ADOS) [117]. Regarding oxidative stress, patients with ASD seem to have higher levels than the general population [119]. This would seem to be mainly due to a reduction in the activity of endogenous antioxidants such as SOD, GTPx, and CAT [120]. The reduction of the enzymatic activity would seem to increase the production of pro-oxidizing and lipid peroxidation-related substances such as F2-isoprostane and 8-iso-prostaglandin F2α [121]. Furthermore, there would seem to be an alteration in metallothionein-3 (MT-3), a protein with detoxifying activity expressed in the brain. This, in fact, seems to be reduced in patients with ASD, causing greater neuronal toxicity [119]. It should also be mentioned that, in general, in patients with ASD, there is a reduction in total glutathione with consequently reduced detoxification of pro-oxidizing metabolites [121].
2.8. Primitive Immunodeficiencies
Primitive immunodeficiencies (PID) represent a wide group of diseases, generally considered as conditions with an increased rate of serious and recurring infections caused by an alteration in the immune response. In the immuno-dysregulated scenario, along with the augmented infection susceptibility, there is also a manifestation of autoimmunity. As said, under the definition of PID, there are numerous clinical syndromes [122]. However, the correlation between these syndromes and oxidative stress has been sufficiently studied only in a few of them, as follows: the common variable immunodeficiency disease (CVID), the most serious subtype of severe combined immunodeficiency (SCID), the reticular dysgenesis (RD), and the chronic granulomatous disease (CGD) [123].
2.9. Gaucher Disease
Gaucher disease (GD) is one of the most common lysosomal storage diseases and it is caused by mutations in the gene GBA1. GBA1 encodes the enzyme glucocerebrosidase (GCase, acid-β-glucosidase) that leads to altered glucocerebrosidase activity, resulting in the accumulation and storage of glycosphingolipids as the glucocerebroside (GL1) in the lysosomes of macrophages. GL1-engorged macrophages (Gaucher cells) are usually found in the bone marrow, spleen, liver, lungs, lymph nodes, and other organs of patients with GD [124][125].
Glucosylsphingosine (GlcSph, lyso-GL1, lyso-Gb1) is another glycosphingolipid that is present in much lower concentration but probably more pathogenic than GL1 itself in patients with GD [126]. There are the following three different subtypes of GD, according to the presence of neurological deterioration, age at identification, and disease progression rate: the most common is represented by GD1 (non-neuronopathic); GD2 (acute neuronopathic), and GD3 (chronic neuronopathic), which are less common but are associated with a more severe phenotype [127]. GD is normally suspected for the presence of unexpected anemia, thrombocytopenia, and organomegaly. The gold standard for the diagnosis of GD is the presence of low enzymatic activity of GBA1 in peripheral blood compared with normal controls [128][129].
There are the following four treatments available for GD1: 3 ERTs and 1 SRT. No drugs have been approved for GD2 or GD3 [124]. Despite the current therapies, chronic pain and fatigue are two of the most important symptoms associated with GD1. This is probably due to the chronic inflammation associated with the release of pro-inflammatory cytokines and other mediators either directly from Gaucher macrophages or indirectly by crosstalk between Gaucher cells and immunomodulatory lymphocytes [130]. However, it has been reported that the accumulation of toxic glycosphingolipids within cells leads to the production of reactive oxygen species (ROS) and an imbalance between the pro-oxidants and the antioxidant reserve, resulting in oxidative stress and inflammation [131].
The loss of the function of GCase is in fact responsible for an important decrease in the mitochondrial membrane potential, adenosine diphosphate phosphorylation, and an increase in oxidative stress and fragmentation of mitochondria [132]. For this reason, different authors have tried to study the relation between oxidative stress and GD. Rollo et al. investigated the relationship between ROS and GD by analyzing blood oxidative stress markers in GD patients submitted to ERT at different stages during the treatment. They discovered that RT-treated GD patients showed an improvement in antioxidant capacity, which was further increased just after recombinant enzyme infusion [133]. Mello et al. showed an alteration in the concentration of CAT, SOD, and SH, which suggests that there was a change in reactive oxygen species in GD type I patients when compared to HC. This increase in CAT, SOD, and sulfhydryl could likely be related to the prevention of the increase in hydrogen peroxide, preventing damage to lipids [131].
Recently, Kartha et al. decided to study the levels of multiple oxidative stress biomarkers in plasma and red blood cells from untreated patients, in stable individuals undergoing standard-of-care therapy, and in healthy controls.. They found significant differences in key oxidative stress biomarkers in untreated patients compared to healthy controls, while in treated patients, results generally fell between the controls and the untreated patients [128].
References
- Finkel, T.; Holbrook, N.J. Oxidants, oxidative stress and the biology of ageing. Nature 2000, 408, 239.
- Sies, H. On the history of oxidative stress: Concept and some aspects of current development. Curr. Opin. Toxicol. 2018, 7, 122–126.
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383.
- Gulcin, İ. Antioxidants and antioxidant methods: An updated overview. Arch. Toxicol. 2020, 94, 651–715.
- Pallardó, F.V.; Lloret, A.; Lebel, M.; D’Ischia, M.; Cogger, V.C.; Le Couteur, D.G.; Gadaleta, M.N.; Castello, G.; Pagano, G. Mitochondrial dysfunction in some oxidative stress-related genetic diseases: Ataxia-Telangiectasia, Down Syndrome, Fanconi Anaemia and Werner Syndrome. Biogerontology 2010, 11, 401–419.
- Fiore, M.; Petrella, C.; Coriale, G.; Rosso, P.; Fico, E.; Ralli, M.; Greco, A.; De Vincentiis, M.; Minni, A.; Polimeni, A.; et al. Markers of Neuroinflammation in the Serum of Prepubertal Children with Fetal Alcohol Spectrum Disorders. CNS Neurol. Disord.—Drug Targets 2021, 20.
- Ferraguti, G.; Merlino, L.; Battagliese, G.; Piccioni, M.G.; Barbaro, G.; Carito, V.; Messina, M.P.; Scalese, B.; Coriale, G.; Fiore, M.; et al. Fetus morphology changes by second-trimester ultrasound in pregnant women drinking alcohol. Addict. Biol. 2020, 25, e12724.
- Coriale, G.; Fiorentino, D.; Lauro, F.D.I.; Marchitelli, R.; Scalese, B.; Fiore, M.; Maviglia, M.; Ceccanti, M. Fetal Alcohol Spectrum Disorder (FASD): Neurobehavioral profile, indications for diagnosis and treatment. Riv. Psichiatr. 2013, 48, 359–369.
- Wilhoit, L.F.; Scott, D.A.; Simecka, B.A. Fetal Alcohol Spectrum Disorders: Characteristics, Complications, and Treatment. Community Ment. Health J. 2017, 53, 711–718.
- Denny, L.A.; Coles, S.; Blitz, R. Fetal Alcohol Syndrome and Fetal Alcohol Spectrum Disorders. Am. Fam. Physician 2017, 96, 515–522.
- Mattson, S.N.; Crocker, N.; Nguyen, T.T. Fetal alcohol spectrum disorders: Neuropsychological and behavioral features. Neuropsychol. Rev. 2011, 21, 81–101.
- Goodlett, C.R.; Horn, K.H.; Zhou, F.C. Alcohol teratogenesis: Mechanisms of damage and strategies for intervention. Exp. Biol. Med. 2005, 230, 394–406.
- Ciafrè, S.; Carito, V.; Ferraguti, G.; Greco, A.; Chaldakov, G.N.; Fiore, M.; Ceccanti, M. How alcohol drinking affects our genes: An epigenetic point of view. Biochem. Cell Biol. 2019, 97, 345–356.
- Lange, S.; Probst, C.; Gmel, G.; Rehm, J.; Burd, L.; Popova, S. Global prevalence of fetal alcohol spectrum disorder among children and youth: A systematic review and meta-analysis. JAMA Pediatr. 2017, 171, 948–956.
- Banakar, M.K.; Kudlur, N.S.; George, S. Fetal alcohol spectrum disorder(FASD. Indian J. Pediatr. 2009, 76, 1173–1175.
- Aragón, A.S.; Coriale, G.; Fiorentino, D.; Kalberg, W.O.; Buckley, D.; Phillip Gossage, J.; Ceccanti, M.; Mitchell, E.R.; May, P.A. Neuropsychological characteristics of Italian children with fetal alcohol spectrum disorders. Alcohol. Clin. Exp. Res. 2008, 32, 1909–1919.
- May, P.A.; Fiorentino, D.; Phillip Gossage, J.; Kalberg, W.O.; Eugene Hoyme, H.; Robinson, L.K.; Coriale, G.; Jones, K.L.; Del Campo, M.; Tarani, L.; et al. Epidemiology of FASD in a province in Italy: Prevalence and characteristics of children in a random sample of schools. Alcohol. Clin. Exp. Res. 2006, 30, 1562–1575.
- Ferraguti, G.; Ciolli, P.; Carito, V.; Battagliese, G.; Mancinelli, R.; Ciafrè, S.; Tirassa, P.; Ciccarelli, R.; Cipriani, A.; Messina, M.P.; et al. Ethylglucuronide in the urine as a marker of alcohol consumption during pregnancy: Comparison with four alcohol screening questionnaires. Toxicol. Lett. 2017, 275, 49–56.
- Ciafrè, S.; Ferraguti, G.; Greco, A.; Polimeni, A.; Ralli, M.; Ceci, F.M.; Ceccanti, M.; Fiore, M. Alcohol as an early life stressor: Epigenetics, metabolic, neuroendocrine and neurobehavioral implications. Neurosci. Biobehav. Rev. 2020, 118, 654–668.
- Abel, E.L.; Sokol, R.J. A Revised Conservative Estimate of the Incidence of FAS and its Economic Impact. Alcohol. Clin. Exp. Res. 1991, 15, 514–524.
- Stade, B.; Ali, A.; Bennett, D.; Campbell, D.; Johnston, M.; Lens, C.; Tran, S.; Koren, G. The burden of prenatal exposure to alcohol: Revised measurement of cost. Can. J. Clin. Pharmacol. 2009, 16, e91–e102.
- Gupta, K.K.; Gupta, V.K.; Shirasaka, T. An Update on Fetal Alcohol Syndrome-Pathogenesis, Risks, and Treatment. Alcohol. Clin. Exp. Res. 2016, 40, 1594–1602.
- Szwajgier, D.; Baranowska-Wójcik, E.; Grzelczyk, J.; Żukiewicz-Sobczak, W. Peripheral Oxidation Markers in Down Syndrome Patients: The Better and the Worse. Dis. Markers 2021, 2021, 1–9.
- Davis-Anderson, K.L.; Wesseling, H.; Siebert, L.M.; Lunde-Young, E.R.; Naik, V.D.; Steen, H.; Ramadoss, J. Fetal regional brain protein signature in FASD rat model. Reprod. Toxicol. 2018, 76, 84–92.
- Bhatia, S.; Drake, D.M.; Miller, L.; Wells, P.G. Oxidative stress and DNA damage in the mechanism of fetal alcohol spectrum disorders. Birth Defects Res. 2019, 111, 714–748.
- Chater-Diehl, E.J.; Laufer, B.I.; Castellani, C.A.; Alberry, B.L.; Singh, S.M. Alteration of gene expression, DNA methylation, and histone methylation in free radical scavenging networks in adult mouse hippocampus following fetal alcohol exposure. PLoS ONE 2016, 11, e0154836.
- Khalid, O.; Kim, J.J.; Kim, H.S.; Hoang, M.; Tu, T.G.; Elie, O.; Lee, C.; Vu, C.; Horvath, S.; Spigelman, I.; et al. Gene expression signatures affected by alcohol-induced DNA methylomic deregulation in human embryonic stem cells. Stem Cell Res. 2014, 12, 791–806.
- Van Houten, B.; Woshner, V.; Santos, J.H. Role of mitochondrial DNA in toxic responses to oxidative stress. DNA Repair 2006, 5, 145–152.
- Memo, L.; Gnoato, E.; Caminiti, S.; Pichini, S.; Tarani, L. Fetal alcohol spectrum disorders and fetal alcohol syndrome: The state of the art and new diagnostic tools. Early Hum. Dev. 2013, 89, S40–S43.
- Brocardo, P.S.; Gil-Mohapel, J.; Christie, B.R. The Role of Oxidative Stress in Fetal Alcohol Spectrum Disorders. Brain Res. Rev. 2011, 67, 209–225.
- Brzezinski, M.R.; Boutelet-Bochan, H.; Person, R.E.; Fantel, A.G.; Juchau, M.R. Catalytic activity and quantitation of cytochrome P-450 2E1 in prenatal human brain. J. Pharmacol. Exp. Ther. 1999, 289, 1648–1653.
- De La Monte, S.M.; Kril, J.J. Human alcohol-related neuropathology. Acta Neuropathol. 2014, 127, 71–90.
- Halliwell, B. Reactive oxygen species in living systems: Source, biochemistry, and role in human disease. Am. J. Med. 1991, 91, S14–S22.
- Walton, J.R.; Martens, M.A.; Pober, B.R. The proceedings of the 15th professional conference on Williams Syndrome. Am. J. Med. Genet. Part A 2017, 173, 1159–1171.
- Bódizs, R.; Gombos, F.; Gerván, P.; Szocs, K.; Réthelyi, J.M.; Kovács, I. Aging and sleep in Williams syndrome: Accelerated sleep deterioration and decelerated slow wave sleep decrement. Res. Dev. Disabil. 2014, 35, 3226–3235.
- Lenhoff, H.M.; Wang, P.P.; Greenberg, F.; Bellugi, U. Williams syndrome and the brain. Sci. Am. 1997, 277, 68–73.
- Wan, E.S.; Pober, B.R.; Washko, G.R.; Raby, B.A.; Silverman, E.K. Pulmonary function and emphysema in Williams-Beuren syndrome. Am. J. Med. Genet. Part A 2010, 152, 653–656.
- Ferrari, M.; Stagi, S. Oxidative stress in down and williams-beuren syndromes: An overview. Molecules 2021, 26, 3139.
- Gavazzi, G.; Faury, G. NOX- and ROS-Driven Hypertension in Elastin Insufficiency. Function 2021, 2, zqab035.
- Kozel, B.A.; Danback, J.R.; Waxler, J.L.; Knutsen, R.H.; De Las Fuentes, L.; Reusz, G.S.; Kis, E.; Bhatt, A.B.; Pober, B.R. Williams syndrome predisposes to vascular stiffness modified by antihypertensive use and copy number changes in NCF1. Hypertension 2014, 63, e74–e80.
- McNally, J.S.; Davis, M.E.; Giddens, D.P.; Saha, A.; Hwang, J.; Dikalov, S.; Jo, H.; Harrison, D.G. Role of xanthine oxidoreductase and NAD(P)H oxidase in endothelial superoxide production in response to oscillatory shear stress. Am. J. Physiol.—Hear. Circ. Physiol. 2003, 285, H2290-H2297.
- Csiszar, A.; Lehoux, S.; Ungvari, Z. Hemodynamic forces, vascular oxidative stress, and regulation of BMP-2/4 expression. Antioxidants Redox Signal. 2009, 11, 1683–1697.
- Lehoux, S. Redox signalling in vascular responses to shear and stretch. Cardiovasc. Res. 2006, 71, 269–279.
- Meroni, S.B.; Galardo, M.N.; Rindone, G.; Gorga, A.; Riera, M.F.; Cigorraga, S.B. Molecular mechanisms and signaling pathways involved in Sertoli cell proliferation. Front. Endocrinol. 2019, 10, 224.
- Troia, A.; Knutsen, R.H.; Halabi, C.M.; Malide, D.; Yu, Z.X.; Wardlaw-Pickett, A.; Kronquist, E.K.; Tsang, K.M.; Kovacs, A.; Mecham, R.P.; et al. Inhibition of NOX1 Mitigates Blood Pressure Increases in Elastin Insufficiency. Function 2021, 2, zqab015.
- Tebbenkamp, A.T.N.; Varela, L.; Choi, J.; Paredes, M.I.; Giani, A.M.; Song, J.E.; Sestan-Pesa, M.; Franjic, D.; Sousa, A.M.M.; Liu, Z.W.; et al. The 7q11.23 Protein DNAJC30 Interacts with ATP Synthase and Links Mitochondria to Brain Development. Cell 2018, 175, 1088–1104.
- Mizumura, K.; Cloonan, S.M.; Nakahira, K.; Bhashyam, A.R.; Cervo, M.; Kitada, T.; Glass, K.; Owen, C.A.; Mahmood, A.; Washko, G.R.; et al. Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J. Clin. Investig. 2014, 124, 3987–4003.
- Pouwels, S.D.; Zijlstra, G.J.; van der Toorn, M.; Hesse, L.; Gras, R.; Hacken, N.H.T.T.; Krysko, D.V.; Vandenabeele, P.; De Vries, M.; Van Oosterhout, A.J.M.; et al. Cigarette smoke-induced necroptosis and DAMP release trigger neutrophilic airway inflammation in mice. Am. J. Physiol.—Lung Cell. Mol. Physiol. 2016, 310, L377-L386.
- Van Der Toorn, M.; Slebos, D.J.; De Bruin, H.G.; Leuvenink, H.G.; Bakker, S.J.L.; Gans, R.O.B.; Koëter, G.H.; Van Oosterhout, A.J.M.; Kauffman, H.F. Cigarette smoke-induced blockade of the mitochondrial respiratory chain switches lung epithelial cell apoptosis into necrosis. Am. J. Physiol.—Lung Cell. Mol. Physiol. 2007, 292, L1156-L1162.
- Rangarajan, S.; Bernard, K.; Thannickal, V.J. Mitochondrial dysfunction in pulmonary fibrosis. Ann. Am. Thorac. Soc. 2017, 14, S383–S388.
- Cadenas, E.; Davies, K.J.A. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 2000, 29, 222–230.
- Lavin, M.F.; Shiloh, Y. Ataxia-telangiectasia: A multifaceted genetic disorder associated with defective signal transduction. Curr. Opin. Immunol. 1996, 8, 459–464.
- Lavin, M.F. Ataxia-telangiectasia: From a rare disorder to a paradigm for cell signalling and cancer. Nat. Rev. Mol. Cell Biol. 2008, 9, 759–769.
- Derheimer, F.A.; Kastan, M.B. Multiple roles of ATM in monitoring and maintaining DNA integrity. FEBS Lett. 2010, 584, 3675–3681.
- Mutant ATM Withdraws Translation. Sci. STKE 2000, 2000, 2000.
- Schubert, R.; Erker, L.; Barlow, C.; Yakushiji, H.; Larson, D.; Russo, A.; Mitchell, J.B.; Wynshaw-Boris, A. Cancer chemoprevention by the antioxidant tempol in Atm-deficient mice. Hum. Mol. Genet. 2004, 13, 1793–1802.
- Liu, N.; Stoica, G.; Yan, M.; Scofield, V.L.; Qiang, W.; Lynn, W.S.; Wong, P.K.Y. ATM deficiency induces oxidative stress and endoplasmic reticulum stress in astrocytes. Lab. Investig. 2005, 85, 1471–1480.
- Stagni, V.; Cirotti, C.; Barilà, D. Ataxia-telangiectasia mutated kinase in the control of oxidative stress, mitochondria, and autophagy in cancer: A maestro with a large orchestra. Front. Oncol. 2018, 8, 73.
- Fang, E.F.; Kassahun, H.; Croteau, D.L.; Scheibye-Knudsen, M.; Marosi, K.; Lu, H.; Shamanna, R.A.; Kalyanasundaram, S.; Bollineni, R.C.; Wilson, M.A.; et al. NAD+ Replenishment Improves Lifespan and Healthspan in Ataxia Telangiectasia Models via Mitophagy and DNA Repair. Cell Metab. 2016, 24, 566–581.
- Weyemia, U.; Redon, C.E.; Aziz, T.; Choudhuri, R.; Maeda, D.; Parekh, P.R.; Bonner, M.Y.; Arbiser, J.L.; Bonner, W.M. NADPH oxidase 4 is a critical mediator in Ataxia telangiectasia disease. Proc. Natl. Acad. Sci. USA 2015, 112, 2121–2126.
- Sies, H. Oxidative stress: Concept and some practical aspects. Antioxidants 2020, 9, 852.
- Guo, S.; Chen, X. The human Nox4: Gene, structure, physiological function and pathological significance. J. Drug Target. 2015, 23, 888–896.
- Maciejczyk, M.; Mikoluc, B.; Pietrucha, B.; Heropolitanska-Pliszka, E.; Pac, M.; Motkowski, R.; Car, H. Oxidative stress, mitochondrial abnormalities and antioxidant defense in Ataxia-telangiectasia, Bloom syndrome and Nijmegen breakage syndrome. Redox Biol. 2017, 11, 375–383.
- Yang, M.; Luna, L.; Sørbø, J.G.; Alseth, I.; Johansen, R.F.; Backe, P.H.; Danbolt, N.C.; Eide, L.; Bjørås, M. Human OXR1 maintains mitochondrial DNA integrity and counteracts hydrogen peroxide-induced oxidative stress by regulating antioxidant pathways involving p21. Free Radic. Biol. Med. 2014, 77, 41–48.
- Semlitsch, M.; Shackelford, R.E.; Zirkl, S.; Sattler, W.; Malle, E. ATM protects against oxidative stress induced by oxidized low-density lipoprotein. DNA Repair 2011, 10, 848–860.
- Matsuura, E.; Kobayashi, K.; Tabuchi, M.; Lopez, L.R. Oxidative modification of low-density lipoprotein and immune regulation of atherosclerosis. Prog. Lipid Res. 2006, 45, 466–486.
- Mazière, C.; Mazière, J.C. Activation of transcription factors and gene expression by oxidized low-density lipoprotein. Free Radic. Biol. Med. 2009, 46, 127–137.
- Shackelford, R.E.; Manuszak, R.P.; Johnson, C.D.; Hellrung, D.J.; Link, C.J.; Wang, S. Iron chelators increase the resistance of Ataxia telangeictasia cells to oxidative stress. DNA Repair 2004, 3, 1263–1272.
- Green, M.H.L.; Marcovitch, A.J.; Harcourt, S.A.; Lowe, J.E.; Green, I.C.; Arlett, C.F. Hypersensitivity of ataxia-telangiectasia fibroblasts to a nitric oxide donor. Free Radic. Biol. Med. 1997, 22, 343–347.
- Patterson, D. Molecular genetic analysis of Down syndrome. Hum. Genet. 2009, 126, 195–214.
- Antonarakis, S.E.; Petersen, M.B.; McInnis, M.G.; Adelsberger, P.A.; Schinzel, A.A.; Binkert, F.; Pangalos, C.; Raoul, O.; Slaugenhaupt, S.A.; Hafez, M.; et al. The meiotic stage of nondisjunction in trisomy 21: Determination by using DNA polymorphisms. Am. J. Hum. Genet. 1992, 50, 544.
- Asim, A.; Kumar, A.; Muthuswamy, S.; Jain, S.; Agarwal, S. Down syndrome: An insight of the disease. J. Biomed. Sci. 2015, 22, 41.
- Lott, I.; Head, E.; Doran, E.; Busciglio, J. Beta-Amyloid, Oxidative Stress and Down Syndrome. Curr. Alzheimer Res. 2006, 3, 521–528.
- Muchova, J.; Zitnanova, I.; Durackova, Z. Oxidative stress and Down syndrome. Do antioxidants play a role in therapy? Physiol. Res. 2014, 63, 535–542.
- Tarani, L.; Carito, V.; Ferraguti, G.; Petrella, C.; Greco, A.; Ralli, M.; Messina, M.P.; Rasio, D.; De Luca, E.; Putotto, C.; et al. Neuroinflammatory Markers in the Serum of Prepubertal Children with down Syndrome. J. Immunol. Res. 2020, 2020, 6937154.
- Lott, I.T.; Head, E. Dementia in Down syndrome: Unique insights for Alzheimer disease research. Nat. Rev. Neurol. 2019, 15, 135–147.
- Perluigi, M.; Tramutola, A.; Pagnotta, S.; Barone, E.; Allan Butterfield, D. The bach1/Nrf2 axis in brain in down syndrome and transition to alzheimer disease-like neuropathology and dementia. Antioxidants 2020, 9, 779.
- Perluigi, M.; Butterfield, D.A. Oxidative stress and down syndrome: A route toward Alzheimer-like dementia. Curr. Gerontol. Geriatr. Res. 2012, 2012, 724904.
- Groner, Y.; Lieman-Hurwitz, J.; Dafni, N.; Sherman, L.; Levanon, D.; Bernstein, Y.; Danciger, E.; Elroy-Stein, O. Molecular Structure and Expression of the Gene Locus on Chromosome 21 Encoding the Cu/Zn Superoxide Dismutase and Its Relevance to Down Syndrome. Ann. N. Y. Acad. Sci. 1985, 450, 133–156.
- Gulesserian, T.; Seidl, R.; Hardmeier, R.; Cairns, N.; Lubec, G. Superoxide dismutase SOD1, encoded on chromosome 21, but not SOD2 is overexpressed in brains of patients with Down Syndrome. J. Investig. Med. 2001, 49, 41–46.
- Barone, E.; Arena, A.; Head, E.; Butterfield, D.A.; Perluigi, M. Disturbance of redox homeostasis in Down Syndrome: Role of iron dysmetabolism. Free Radic. Biol. Med. 2018, 114, 84–93.
- Zana, M.; Janka, Z.; Kálmán, J. Oxidative stress: A bridge between Down’s syndrome and Alzheimer’s disease. Neurobiol. Aging 2007, 28, 648–676.
- De Haan, J.B.; Cristiano, F.; Iannello, R.C.; Kola, I. Cu/Zn-superoxide dismutase and glutathione peroxidase during aging. Biochem. Mol. Biol. Int. 1995, 35, 1281–1297.
- Perluigi, M.; Di Domenico, F.; Buttterfield, D.A. Unraveling the complexity of neurodegeneration in brains of subjects with Down syndrome: Insights from proteomics. Proteomics—Clin. Appl. 2014, 8, 73–85.
- Ma, K.; Li, F.; Yu, Y.; Li, H. Screening of potential biomarkers for prenatal diagnosis of trisomy 21. J. Obstet. Gynaecol. 2017, 37, 435–440.
- Barone, E.; Head, E.; Butterfield, D.A.; Perluigi, M. HNE-modified proteins in Down syndrome: Involvement in development of Alzheimer disease neuropathology. Free Radic. Biol. Med. 2017, 111, 262–269.
- Di Domenico, F.; Pupo, G.; Mancuso, C.; Barone, E.; Paolini, F.; Arena, A.; Blarzino, C.; Schmitt, F.A.; Head, E.; Butterfield, D.A.; et al. Bach1 overexpression in down syndrome correlates with the alteration of the HO-1/BVR-A system: Insights for transition to alzheimer’s disease. J. Alzheimer’s Dis. 2015, 44, 1107–1120.
- Donato, R. Intracellular and extracellular roles of S100 proteins. Microsc. Res. Tech. 2003, 60, 540–551.
- Donato, R.; Sorci, G.; Riuzzi, F.; Arcuri, C.; Bianchi, R.; Brozzi, F.; Tubaro, C.; Giambanco, I. S100B’s double life: Intracellular regulator and extracellular signal. Biochim. Biophys. Acta—Mol. Cell Res. 2009, 1793, 408–415.
- Helguera, P.; Pelsman, A.; Pigino, G.; Wolvetang, E.; Head, E.; Busciglio, J. ets-2 promotes the activation of a mitochondrial death pathway in down’s syndrome neurons. J. Neurosci. 2005, 25, 2295–2303.
- Conti, A.; Fabbrini, F.; D’Agostino, P.; Negri, R.; Greco, D.; Genesio, R.; D’Armiento, M.; Olla, C.; Paladini, D.; Zannini, M.; et al. Altered expression of mitochondrial and extracellular matrix genes in the heart of human fetuses with chromosome 21 trisomy. BMC Genom. 2007, 8, 1–15.
- Yuan, S.M.; Jing, H. Marfan’s syndrome: An overview. Sao Paulo Med. J. 2010, 128, 360–366.
- Dietz, H.C.; Loeys, B.; Carta, L.; Ramirez, F. Recent progress towards a molecular understanding of Marfan syndrome. Am. J. Med. Genet.—Semin. Med. Genet. 2005, 139C, 4–9.
- Sakai, L.Y.; Keene, D.R.; Renard, M.; De Backer, J. FBN1: The disease-causing gene for Marfan syndrome and other genetic disorders. Gene 2016, 592, 279–291.
- Coelho, S.G.; Almeida, A.G. Marfan syndrome revisited: From genetics to the clinic. Rev. Port. Cardiol. 2020, 39, 215–226.
- Zeigler, S.M.; Sloan, B.; Jones, J.A. Pathophysiology and Pathogenesis of Marfan Syndrome. Adv. Exp. Med. Biol. 2021, 1348.
- Hugar, B.S.; Praveen, S.; Kainoor, S.K.; Shetty, A.R.S. Sudden death in marfan syndrome. J. Forensic Sci. 2014, 59, 1126–1128.
- Loeys, B.L.; Dietz, H.C.; Braverman, A.C.; Callewaert, B.L.; De Backer, J.; Devereux, R.B.; Hilhorst-Hofstee, Y.; Jondeau, G.; Faivre, L.; Milewicz, D.M.; et al. The revised Ghent nosology for the Marfan syndrome. J. Med. Genet. 2010, 47, 530–532.
- Griendling, K.K.; Sorescu, D.; Ushio-Fukai, M. NAD(P)H oxidase: Role in cardiovascular biology and disease. Circ. Res. 2000, 86, 494–501.
- Owens, G.K.; Kumar, M.S.; Wamhoff, B.R. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol. Rev. 2004, 84, 767–801.
- Yang, H.H.C.; van Breemen, C.; Chung, A.W.Y. Vasomotor dysfunction in the thoracic aorta of Marfan syndrome is associated with accumulation of oxidative stress. Vascul. Pharmacol. 2010, 52, 37–45.
- Liaw, N.; Dolan Fox, J.M.; Siddiqui, A.H.; Meng, H.; Kolega, J. Endothelial nitric oxide synthase and superoxide mediate hemodynamic initiation of intracranial aneurysms. PLoS ONE 2014, 9, e101721.
- Rysz, J.; Gluba-Brzózka, A.; Rokicki, R.; Franczyk, B. Oxidative stress-related susceptibility to aneurysm in marfan’s syndrome. Biomedicines 2021, 9, 1171.
- Jiménez-Altayó, F.; Meirelles, T.; Crosas-Molist, E.; Sorolla, M.A.; del Blanco, D.G.; López-Luque, J.; Mas-Stachurska, A.; Siegert, A.M.; Bonorino, F.; Barberà, L.; et al. Redox stress in Marfan syndrome: Dissecting the role of the NADPH oxidase NOX4 in aortic aneurysm. Free Radic. Biol. Med. 2018, 118, 44–58.
- Chung, A.W.Y.; Yang, H.H.C.; Au Yeung, K.; Van Breemen, C. Mechanical and pharmacological approaches to investigate the pathogenesis of marfan syndrome in the abdominal aorta. J. Vasc. Res. 2008, 45, 314–322.
- Fiorillo, C.; Becatti, M.; Attanasio, M.; Lucarini, L.; Nassi, N.; Evangelisti, L.; Porciani, M.C.; Nassi, P.; Gensini, G.F.; Abbate, R.; et al. Evidence for oxidative stress in plasma of patients with Marfan syndrome. Int. J. Cardiol. 2010, 145, 544–546.
- Tsui, V.; Crismani, W. The Fanconi Anemia Pathway and Fertility. Trends Genet. 2019, 35, 199–214.
- Liu, W.; Palovcak, A.; Li, F.; Zafar, A.; Yuan, F.; Zhang, Y. Fanconi anemia pathway as a prospective target for cancer intervention. Cell Biosci. 2020, 10, 1–14.
- Kulanuwat, S.; Jungtrakoon, P.; Tangjittipokin, W.; Yenchitsomanus, P.T.; Plengvidhya, N. Fanconi anemia complementation group C protection against oxidative stress-induced ß-cell apoptosis. Mol. Med. Rep. 2018, 18, 2485–2491.
- Solomon, B.D. VACTERL/VATER association. Orphanet J. Rare Dis. 2011, 6, 56.
- Niraj, J.; Färkkilä, A.; D’Andrea, A.D. The fanconi anemia pathway in cancer. Annu. Rev. Cancer Biol. 2019, 3, 2585–2588.
- Palovcak, A.; Liu, W.; Yuan, F.; Zhang, Y. Maintenance of genome stability by Fanconi anemia proteins. Cell Biosci. 2017, 7, 1–18.
- Du, W.; Adam, Z.; Rani, R.; Zhang, X.; Pang, Q. Oxidative stress in fanconi anemia hematopoiesis and disease progression. Antioxidants Redox Signal. 2008, 10, 1909–1921.
- Pagano, G.; Talamanca, A.A.; Castello, G.; Pallardó, F.V.; Zatterale, A.; Degan, P. Oxidative stress in Fanconi anaemia: From cells and molecules towards prospects in clinical management. Biol. Chem. 2012, 393, 11–21.
- Reiss, A.L. Childhood developmental disorders: An academic and clinical convergence point for psychiatry, neurology, psychology and pediatrics. J. Child Psychol. Psychiatry Allied Discip. 2009, 50, 87–98.
- Courchesne, E.; Gazestani, V.H.; Lewis, N.E. Prenatal Origins of ASD: The When, What, and How of ASD Development. Trends Neurosci. 2020, 43, 326–342.
- Rosen, N.E.; Lord, C.; Volkmar, F.R. The Diagnosis of Autism: From Kanner to DSM-III to DSM-5 and Beyond. J. Autism Dev. Disord. 2021, 51, 4253–4270.
- Manivasagam, T.; Arunadevi, S.; Essa, M.M.; SaravanaBabu, C.; Borah, A.; Thenmozhi, A.J.; Qoronfleh, M.W. Role of Oxidative Stress and Antioxidants in Autism. Adv. Neurobiol. 2020, 24.
- Bjørklund, G.; Meguid, N.A.; El-Bana, M.A.; Tinkov, A.A.; Saad, K.; Dadar, M.; Hemimi, M.; Skalny, A.V.; Hosnedlová, B.; Kizek, R.; et al. Oxidative Stress in Autism Spectrum Disorder. Mol. Neurobiol. 2020, 57, 2314–2332.
- Liu, X.; Lin, J.; Zhang, H.; Khan, N.U.; Zhang, J.; Tang, X.; Cao, X.; Shen, L. Oxidative Stress in Autism Spectrum Disorder—Current Progress of Mechanisms and Biomarkers. Front. Psychiatry 2022, 13, 162.
- Chauhan, A.; Chauhan, V. Oxidative stress in autism. Pathophysiology 2006, 13, 171–181.
- Maffeis, M.; Notarangelo, L.D.; Schumacher, R.F.; Soncini, E.; Soresina, A.; Lanfranchi, A.; Porta, F. Primary immunodeficiencies and oncological risk: The experience of the Children’s Hospital of Brescia. Front. Pediatr. 2019, 7, 232.
- Xiao, X.; Miao, Q.; Chang, C.; Gershwin, M.E.; Ma, X. Common variable immunodeficiency and autoimmunity—An inconvenient truth. Autoimmun. Rev. 2014, 13, 858–864.
- Bennett, L.L.; Mohan, D. Gaucher disease and its treatment options. Ann. Pharmacother. 2013, 47, 1182–1193.
- Burrow, T.A.; Sun, Y.; Prada, C.E.; Bailey, L.; Zhang, W.; Brewer, A.; Wu, S.W.; Setchell, K.D.R.; Witte, D.; Cohen, M.B.; et al. CNS, lung, and lymph node involvement in Gaucher disease type 3 after 11years of therapy: Clinical, histopathologic, and biochemical findings. Mol. Genet. Metab. 2015, 114, 233–241.
- Sidransky, E. Gaucher disease: Complexity in a “simple” disorder. Mol. Genet. Metab. 2004, 83, 6–15.
- Lee, J.Y.; Lee, B.H.; Kim, G.H.; Jung, C.W.; Lee, J.; Choi, J.H.; Yoo, H.W. Clinical and genetic characteristics of Gaucher disease according to phenotypic subgroups. Korean J. Pediatr. 2012, 55, 48.
- Kartha, R.V.; Terluk, M.R.; Brown, R.; Travis, A.; Mishra, U.R.; Rudser, K.; Lau, H.; Jarnes, J.R.; Cloyd, J.C.; Weinreb, N.J. Patients with Gaucher disease display systemic oxidative stress dependent on therapy status. Mol. Genet. Metab. Reports 2020, 25, 100667.
- Zimran, A.; Elstein, D. Gaucher Disease and Related Lysosomal Storage Diseases. In Williams Hematology, 9th ed.; McGraw-Hill Education: New York, NY, USA, 2015.
- Gervas-Arruga, J.; Cebolla, J.J.; De Blas, I.; Roca, M.; Pocovi, M.; Giraldo, P. The influence of genetic variability and proinflammatory status on the development of bone disease in patients with Gaucher disease. PLoS ONE 2015, 10, e0126153.
- Mello, A.S.; Da Silva Garcia, C.; De Souza Machado, F.; Da Silva Medeiros, N.; Wohlenberg, M.F.; Marinho, J.P.; Dani, C.; Funchal, C.; Coelho, J.C. Oxidative stress parameters of Gaucher disease type I patients. Mol. Genet. Metab. Rep. 2015, 4, 1–5.
- Cleeter, M.W.J.; Chau, K.Y.; Gluck, C.; Mehta, A.; Hughes, D.A.; Duchen, M.; Wood, N.W.; Hardy, J.; Mark Cooper, J.; Schapira, A.H. Glucocerebrosidase inhibition causes mitochondrial dysfunction and free radical damage. Neurochem. Int. 2013, 62, 1–7.
- Roversi, F.M.; Galdieri, L.C.; Grego, B.H.C.; Souza, F.G.; Micheletti, C.; Martins, A.M.; D’Almeida, V. Blood oxidative stress markers in Gaucher disease patients. Clin. Chim. Acta 2006, 364, 316–320.
More
Information
Subjects:
Pediatrics
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.1K
Revisions:
2 times
(View History)
Update Date:
01 Nov 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No