Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Qiang Sun | -- | 4943 | 2022-10-27 08:26:35 | | | |
| 2 | Conner Chen | -1 word(s) | 4942 | 2022-10-27 08:37:43 | | | | |
| 3 | Conner Chen | Meta information modification | 4942 | 2022-11-01 01:19:45 | | | | |
| 4 | Conner Chen | Meta information modification | 4942 | 2022-11-01 01:23:07 | | | | |
| 5 | Conner Chen | Meta information modification | 4942 | 2022-11-01 01:29:18 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Sun, Q. The Hydrophobic Effects. Encyclopedia. Available online: https://encyclopedia.pub/entry/31528 (accessed on 23 July 2026).
Sun Q. The Hydrophobic Effects. Encyclopedia. Available at: https://encyclopedia.pub/entry/31528. Accessed July 23, 2026.
Sun, Qiang. "The Hydrophobic Effects" Encyclopedia, https://encyclopedia.pub/entry/31528 (accessed July 23, 2026).
Sun, Q. (2022, October 27). The Hydrophobic Effects. In Encyclopedia. https://encyclopedia.pub/entry/31528
Sun, Qiang. "The Hydrophobic Effects." Encyclopedia. Web. 27 October, 2022.
Copy Citation
Hydrophobic interactions are involved in and believed to be the fundamental driving force of many chemical and biological phenomena in aqueous environments.
water
solute
interface
hydrogen bonding
hydrophobic effects
1. Introduction
Hydrophobic effects refer to the tendency of nonpolar molecules (or parts of molecules) to be aggregated in water. They are involved in and believed to be the fundamental driving force in many chemical and biological phenomena in aqueous solutions, such as molecular recognition, protein folding, formation and stability of micelles, biological membranes and macromolecular complexes, surfactant aggregation, coagulation, complexation, detergency, and the formation of gas clathrates [1][2][3][4][5]. To date, numerous experimental and theoretical works have been carried out to understand the physical origin of hydrophobic effects.
In general, hydrophobicity is expressed through the empirically calculated logarithm of partition coefficient (logP), which is widely used in drug design and medicinal chemistry [6][7]. Historically, the concept of hydrophobicity arose in the context of the low solubility of non-polar solutes in water. Experimentally, it has been found that the entropy change upon transferring a hydrocarbon from a nonpolar environment into water is large and negative. In 1945, Frank and Evans [8] suggested that the observed loss of entropy was related to the structural changes of liquid water as hydrophobic solutes were dissolved into water. They proposed that a kind of “cage”, consisting of water molecules, was formed around the solute. This ordered water structure, similar to the gas clathrate, was known as the “iceberg” model [8]. Since then, many works [9][10][11][12][13][14][15][16][17] are conducted to measure the water structural changes around hydrophobic surfaces. In accordance with the “iceberg” model, some studies [9][10][11][12] support the existence of increased tetrahedral order around small hydrophobic groups in aqueous solutions. However, the decrease of water structure around hydrophobic groups is also found in many works [13][14][15][16][17]. It is well known that the neutron scattering may be sensitive to the structure of water. From the neutron scattering experimental measurements on aqueous solutions containing tetramethylammonium chloride [13] and methane molecules [15], these do not suggest that water around these hydrophobic solutes may be more ordered than bulk water. To date, there remain strong debates on the “iceberg” structural model.
In 1959, based on the “iceberg” model, Kauzmann [18] introduced the concept of hydrophobic interactions. When two “caged” hydrophobes come together, the “structured” water between solutes may be released into the bulk (Figure 1), which undoubtedly leads to the increase of entropy. Subsequently, the attractive force between these particles may be related to the entropy increase. Therefore, hydrophobic interactions are classically regarded to be entropy driven. However, the “classic” hydrophobic effects may be in contrast with some works. In Diederich et al. [19] work, they found that complexation of benzene in a cyclophane host molecule was enthalpy driven at room temperature, which was also accompanied with a slightly negative entropy change. Additionally, in Baron, Setny, and McCammon works [20][21], molecular dynamics (MD) simulations are used to investigate the binding in a hydrophobic receptor-ligand system. It is found that the association between the non-polar ligand and binding pocket may be driven by enthalpy and opposed by entropy. The “non-classic” hydrophobic effects are ascribed to the release of weakly hydrogen-bonded water molecules into the more strongly hydrogen-bonded bulk water [22].
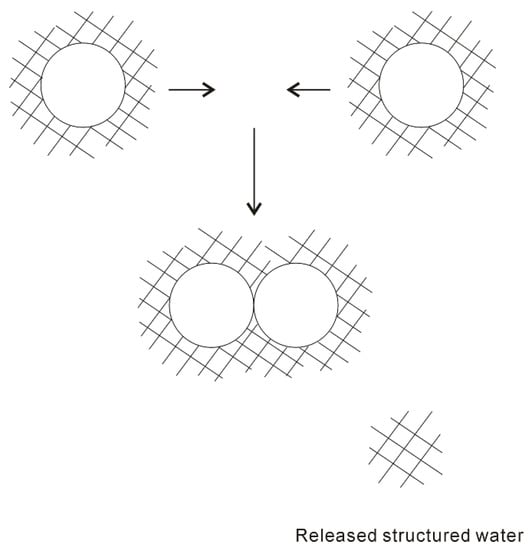
Figure 1. The “iceberg” structural model of hydrophobic effects. The ordered water structure is expected to form around the solute. As two “caged” solutes become together, the “structured” water in the region between them may be returned to the bulk.
In the “iceberg” structural model, water molecules can rearrange around a small hydrophobic solute, without losing their hydrogen bondings. However, as a large solute is embedded into water, hydrogen bondings of water may be broken at the surface of the solute, which may result in an enthalpic penalty. Therefore, hydrophobic interactions depend on the size of dissolved solute. In recent years, a theoretical approach was developed by Lum, Chandler, and Weeks (LCW) [23][24][25][26] to understand the dependence of hydrophobic interactions on solute size. Both the Gaussian density fluctuations related to small size and the physics of interfacial formation related to large size are incorporated in LCW theory [23][24][25][26]. It can be found that the hydration free energy grows linearly with the solvated volume for small solute, but grows linearly with the solvated surface area for large solute [25]. Therefore, the crossover may be expected between small and large regime, which takes place on the nanometer length scale [25].
Liquid water is generally believed to play a vital role in the process of hydrophobic interactions. In cell biology, water may be regarded as an active constituent, rather than a bystander [27][28][29]. According to some recent structural works [30][31][32][33][34][35][36][37][38] on liquid water and air-water interface, hydration free energy is determined. This is utilized to understand the nature of hydrophobic interactions. It is found that hydrophobic interactions may be related to the size of dissolved solute. With increasing the solute size, it is reasonably divided into initial and hydrophobic solvation processes [34]. Additionally, different dissolved behaviors of solutes are expected in initial and hydrophobic solvation processes, such as dispersed and accumulated distributions in aqueous environments. In addition, hydrophobic interactions may be ascribed to the structural competition between interfacial and bulk water [34].
2. Water Structure
Gibbs free energy (ΔG), related to the changes of enthalpy (ΔH) and entropy (ΔS), may be used to study whether a process is likely to take place. Thermodynamically, it is expressed as,
where ΔH is enthalpy changes, and ΔS is entropy changes. In general, ΔH quantifies the average potential energy between molecules, ΔS measures the order (or intermolecular) correlations of system.
In fact, various interactions between solutes and water may be expected when solutes are embedded into water. The total Gibbs free energy of system is reasonably described as,
in which ΔGWater-water, ΔGSolute-water, and ΔGSolute-solute, respectively, mean the Gibbs energies are due to water-water, solute-water, and solute-solute interactions. In fact, the solutes are necessarily attracted to approach each other in aqueous solutions before they may be affected by the interactions between them. This is due to the hydrophobic interactions, which accumulate the solutes in the solutions. Therefore, to understand the molecular mechanism of hydrophobic effects, it is important to study the water structure and the effects of solutes on water structure.
Numerous experimental and theoretical works have been carried out to investigate the structure of water. To date, different structural models have been proposed, which are generally partitioned into the mixture and continuum structural models [39][40]. In the mixture model, two distinct types of structures are regarded to simultaneously exist in ambient water. It is likely that the first mixture model was proposed by W.C. Röntgen in 1892, who suggested that liquid water was a mixture of two components, a low-density fluid and a high-density fluid [41]. Since then, various mixture structural models have been proposed [42][43][44]. For the continuum structural model, water is comprised of a random, three-dimensional hydrogen-bonded network, which may be characterized by a broad distribution of O-H···O hydrogen bond distances and angles. However, the water networks cannot be “broken” (or separated into distinct molecular species) as in the mixture model. To date, liquid water is usually regarded as a tetrahedral fluid, which is based on the first coordination number.
Where ρ means the density of water, rmin and rmax are the lower and upper limits of integration in oxygen-oxygen radial distribution function, gOO(r). For ambient water, Nc is determined to be 4.3 [45] and 4.7 [46], respectively.
Liquid water is usually regarded as an anomalous fluid, which is due to the formation of hydrogen bondings between neighboring water molecules. To understand the physical nature of hydrogen bondings, various theoretical methods have been developed, such as symmetry-adapted perturbation theory (SAPT) [47][48]. From the theoretical calculations [49] on a water dimer ((H2O)2), besides van der Waals interactions between water molecules, obvious electrostatic interactions can also be found between them. Of course, this is reasonably attributed to the formation of hydrogen bondings between water molecules. Therefore, hydrogen bondings may be ascribed to the electrostatic interactions between the neighboring water molecules.
For an H2O molecule, the vibrational normal modes may be 2A1+B1 [50], which are all Raman active. When hydrogen bonding is formed between neighboring water molecules, this decreases the O···O distance between them, and weakens the O-H covalent bond [51]. The formation of hydrogen bonding may result in OH vibrational frequencies moving towards a low wavenumber (red shift). Therefore, OH vibrations may be sensitive to hydrogen bondings of liquid water, and widely utilized to investigate the structure of water.
With decreasing temperature from 298 K to 248 K at 0.1 MPa, based on the normalized intensity, an isosbestic point is found around 3330 cm−1 in the Raman OH stretching bands of water (Figure 2). The isosbestic point is the wavelength where a series of spectra cross, and the spectral intensity may keep constant. In mixture structural model, the isosbestic point is generally used to support the two-state behavior of water structure. However, after considering the electric field experienced by the proton projected onto the OH covalent bond, Smith et al. [52] suggested that the isosbestic point was explained through a continuous distribution of local hydrogen-bonded networks, which was due to the increasing distortions around a single-component tetrahedral structural motif. It is noted that, along with the isosbestic point, temperature increase (or adding NaCl) may also lead to the decrease of the second peak at 4.5 Å in gOO(r), which undoubtedly means the breakage of tetrahedral hydrogen bondings of water. Therefore, it can be derived that the isosbestic point indicates the structural transition between tetrahedral and non-tetrahedral hydrogen-bonded networks.
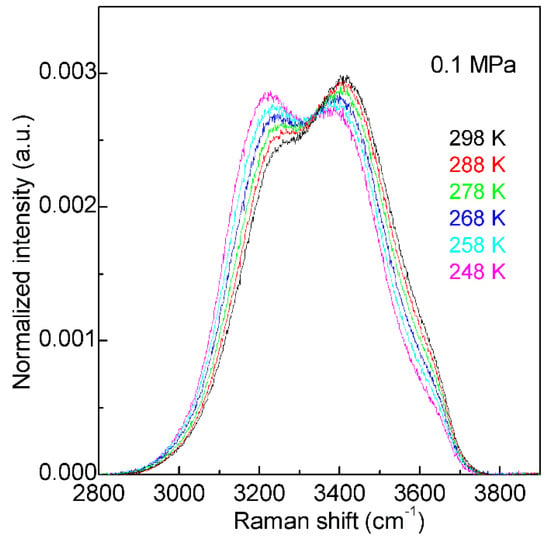
Figure 2. The Raman OH stretching bands of water from 298 to 248 K under 0.1 MPa. Based on normalized intensity, an isosbestic point is found around 3330 cm−1.
Many works have been conducted to explain the Raman OH stretching band of water. In fact, water molecular clusters, (H2O)n, provide an approach to investigate the dependence of OH vibrational frequencies on hydrogen bondings between water molecules. From the theoretical calculations, the stable configurations of water molecular clusters can be determined. In combination with the experimental measurements on OH vibration frequencies of clusters, these may be utilized to unravel the relationship between OH vibrational frequencies and hydrogen-bonded networks. It is found that, when three-dimensional hydrogen-bonded networks occur in water molecular clusters (n ≥ 6), different OH vibrational frequencies may correspond to various hydrogen bondings in the first shell of a water molecule (local hydrogen bondings), and the effects of hydrogen bondings beyond the first shell on OH vibrational frequencies may be weak (Figure 3). From this, as three-dimensional hydrogen bondings appear, different OH vibrations may be ascribed to OH vibrations engaged into various local hydrogen bondings [30][32].
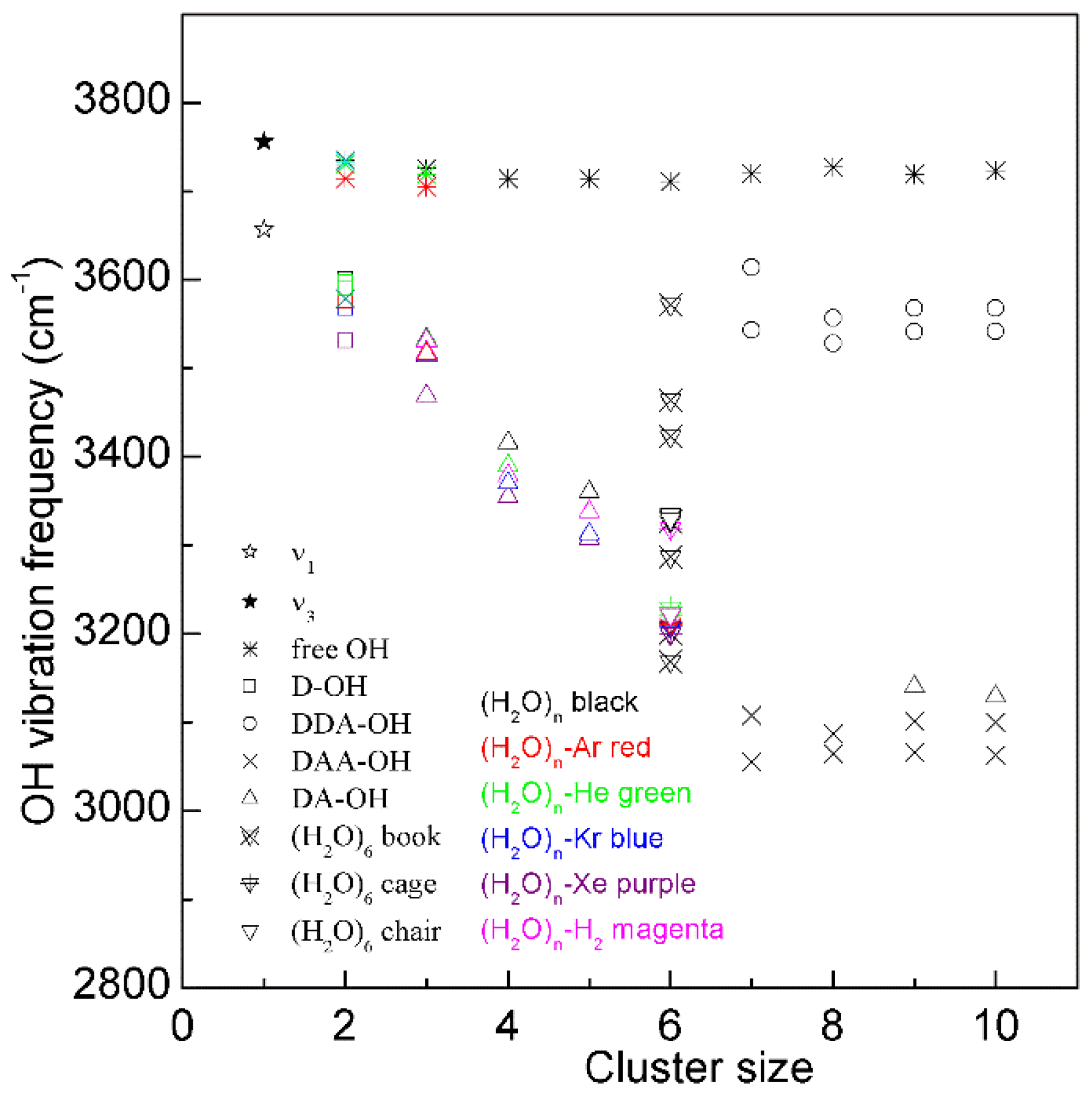
Figure 3. The dependence of the OH stretching frequency on hydrogen bondings of water molecular clusters, (H2O)n. Different symbols are used to discriminate OH vibrations engaged in various local hydrogen-bonded networks of a water molecule. Various structures of hexamers are also shown.
For a water molecule, the local hydrogen-bonded network can be differentiated by whether the molecule forms hydrogen bonds as a proton donor (D), proton acceptor (A), or a combination of both with neighboring molecules. Under ambient conditions, the main local hydrogen bonding motifs for a water molecule can be classified as DDAA (double donor-double acceptor), DDA (double donor-single acceptor), DAA (single donor-double acceptor), and DA (single donor-single acceptor) (Figure 4). For ambient water, the Raman OH stretching band may be fitted into five sub-bands, which can be assigned to the νDAA-OH, νDDAA-OH, νDA-OH, νDDA-OH, and free OH symmetric stretching vibrations, respectively (Figure 4). Therefore, at ambient conditions, different local hydrogen bondings may be expected around a water molecule.
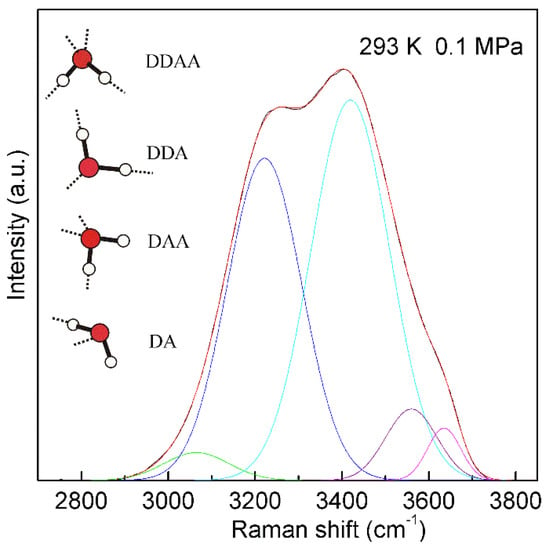
Figure 4. The Raman OH stretching band of ambient water may be deconvoluted into five sub-bands, located at 3041, 3220, 3430, 3572, and 3636 cm−1, and assigned to the νDAA-OH, νDDAA-OH, νDA-OH, νDDA-OH, and free OH symmetric stretching vibrations, respectively. At ambient conditions, the main local hydrogen-bonded networks for a water molecule are expected to be DDAA, DDA, DAA, and DA hydrogen bondings. Hydrogen bondings are drawn with dashed lines.
From the Raman spectroscopic studies [30][31][32] on ambient water, the local statistical model (LSM) is proposed. This suggests that a water molecule interacts with the neighboring water molecules (in the first shell) through different local hydrogen bondings. Of course, it is different from continuum structural models of ambient water. Additionally, according to the mixture structural model, water has been considered as a mixture of two distinct types of structures. Therefore, different spatial distributions may be necessary for various structural types, and sharp boundary is expected between them. In mixture model, water structure may be heterogeneous. Additionally, according to recent X-ray experimental studies [53][54], these mean that liquid water is heterogeneous at ambient conditions, and this becomes enhanced in the supercooled region. However, based on LSM of water structure, various local hydrogen bondings are expected for a water molecule at ambient conditions. This indicates that it is impossible to find a sharp phase boundary between various structural motifs, and the structure of ambient water may be homogeneous. Of course, it is different from the mixture structural model. In addition, based on LSM model, the local hydrogen-bonded networks of a water molecule may be affected by pressure, temperature, dissolved salt, and a confined environment.
According to the explanation on Raman OH stretching band of water [30][31][32], νDDAA-OH is due to OH vibration engaged in DDAA (tetrahedral or “unbroken”) hydrogen bonding, and νFree-OH is free OH symmetric stretching vibration. From the van’t Hoff equation, this may be applied to calculate the thermodynamic functions of tetrahedral (DDAA) hydrogen bonding (Figure 5),
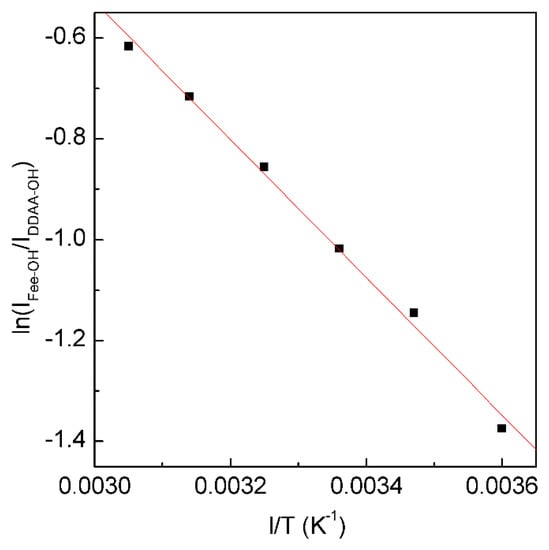
Figure 5. The dependence of ln(IFree-OH/IDDAA-OH) on 1/T. The solid line represents linear fit (R2 = 0.9975) with a slope of −∆H/R. This is used to determine the thermodynamic characteristics of tetrahedral hydrogen bonding.
From the Raman OH stretching bands from 298 K to 248 K, the enthalpy (∆H) and entropy (∆S) from tetrahedral hydrogen bonding to free water are calculated to be 11.35 kJ/mol and 29.66 J/mol, respectively.
Additionally, based on the structural explanation on the Raman OH stretching band of water, it is derived that the isosbestic point (Figure 2) may indicate the structural equilibrium between different local hydrogen bondings around a water molecule. This is expressed as follows,
With decreasing temperature from 298 to 248 K at 0.1 MPa, this decreases the intensity of the high wavenumber sub-bands (>3330 cm−1), but increases the intensity of the low wavenumber sub-bands (<3330 cm−1) (Figure 2). Therefore, temperature decrease may enhance the probability to form tetrahedral hydrogen-bonded networks around a water molecule, which is related to the nucleation of ice. In fact, according to recent MD simulations [55][56][57] on homogeneous ice nucleation, the ordered ice-like intermediate can be found in the supercooled water, and ice nucleation is found to occur in the low-mobility regions [56][57]. Additionally, this intermediate phase has also been found in other theoretical simulations [58][59][60] during the nucleation from supercooled liquids, such as Lenard-Jones hard spheres and metals. From these, a non-classical pathway, rather than classical nucleation theory (CNT), is proposed in order to understand the nucleation mechanism in supercooled liquids.
Water has many unusual thermodynamic and dynamic properties, both in pure form and as a solvent. Additionally, these anomalous behaviors may be strongly enhanced in the supercooled state, such as thermal expansion, isothermal compressibility, etc. To explain the origin of the anomalous behaviors of supercooled water, many theories are proposed, such as the stability limit (SL) conjecture [61], the liquid-liquid critical-point (LLCP) hypothesis [62], the singularity-free (SF) model [63], and the critical-point free scenario [64]. Recently, numerous works [54][65][66][67][68][69] have been carried out to demonstrate the existence of second critical point in LLCP. In fact, LLCP is based on the mixture model of water structure. In LLCP, two liquid phases are expected in water, such as low-density water (LDW) and high-density water (HDW), which may interconvert through a first-order liquid-liquid transition terminating at the second critical point in the supercooled regime. In this hypothesis, the anomalous behavior of water is due to the fluctuations emanating from the LLCP.
With decreasing temperature from 298 to 248 K at 0.1 MPa (Figure 2), this increases the probability to form the tetrahedral (DDAA) hydrogen bondings in supercooled water. In comparison with bulk water, DDAA hydrogen bondings have a larger volume, and lower entropy [32]. This indicates that the formation of tetrahedral hydrogen bondings in a supercooled regime may be used to explain the anomalies of supercooled water, such as the thermal expansion. From statistical mechanics, the thermal expansion may be related to the correlation of the fluctuations of volume and entropy.
For most fluids, with increasing volume, it is also accompanied with an increase of entropy. However, regarding water below TM (temperature of maximum density), the fluctuations of volume and entropy may be anti-correlated. From the above, it is more reasonable to explain the anomalous properties of supercooled water from the SF model [63]. Further study is necessary.
When a solute is dissolved into liquid water, an interface appears between the solute and water. Therefore, the dissolved solute undoubtedly affects the structure of water. At ambient conditions, the OH vibration is mainly related to the local hydrogen-bonded networks of a water molecule. It is derived that the dissolved solute may mainly affect the structure of topmost water layer at the solute-water interface (interfacial water) (Figure 6). In fact, this is in accordance with other studies [70][71][72][73][74] on the structure and dynamics of water around ions. These mean that the effects of dissolved ions on water structure may be largely limited to the first solvation shell. Therefore, as a solute is embedded into water, it may be divided into interfacial and bulk water (Figure 6). Regarding the effects of solutes on water structure, these may be related to the solute-water interfaces. In other words, it is related to the water in confined environments.
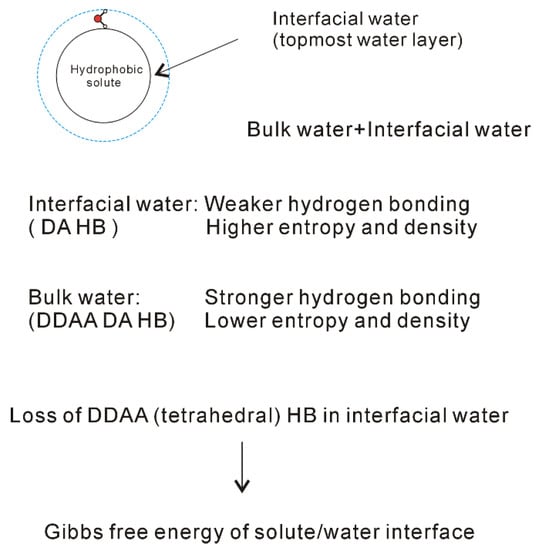
Figure 6. Structural changes across the solute-water interface. The dissolved solute mainly affects the structure of interfacial water (topmost water layer at the interface).
From the Raman spectroscopic studies [30][31][32], DDAA (tetrahedral) and DA are the predominant hydrogen-bonded networks in ambient water. Additionally, they are related to the structural changes across the solute-water interface (Figure 6). It is necessary to understand the characteristics of DDAA and DA hydrogen bondings. For ambient water, the DDAA-OH, located around 3220 cm−1, lies at a lower frequency than DA-OH (3430 cm−1) (Figure 4). This indicates that, in comparison with DA hydrogen bonding, the formation of DDAA structural motif may result in higher hydrogen bonding energies. Additionally, it is necessary for the interacting molecules to lie in specific relative orientations to form hydrogen bondings between neighboring water molecules, therefore DDAA (tetrahedral) is expected to own the lower entropy rather than DA hydrogen bonding. In addition, based on the experimental measurements [75] and theoretical simulations [76][77], higher water density can be found at the solute-water interface, which may be related to the formation of DA in interfacial water. Therefore, higher density is expected for DA as a structural motif than it is for DDAA hydrogen bonding. In comparison with DDAA (tetrahedral) hydrogen-bonded network, the DA structural motif owns a lower enthalpy, and a higher entropy and density.
The dissolved solute mainly affects the structure of interfacial water. Additionally, DA structural motif tends to form at the interface between solute and water. In other words, this means that the loss of tetrahedral hydrogen bonding may be related to the formation of solute-water interface (Figure 6). Certainly, this is different from the “iceberg” structural model proposed by Frank and Evans [8]. In history, the “iceberg” structural model was proposed in order to understand the large and negative entropy while the simple solutes were dissolved in water. In Frank and Evans’ work [8], an increase in the structure of water was utilized to explain the negative entropy incurred from the dissolved non-polar molecules. However, they did not explain why the hydrophobic solute could lead to increase of the order of the system. In fact, this may be due to the transition from interfacial to bulk water as the solutes are dissolved in solutions, which leads to the increase of DDAA hydrogen bondings in water.
The effects of dissolved solute on water structure are mainly limited within the interfacial water layer (Figure 6). Additionally, the formation of solute-water interface is due to the loss of tetrahedral hydrogen bonding in interfacial water. Therefore, as the ratio of interfacial water layer to volume is obtained, this may be utilized to calculate the Gibbs free energy of the interface between solute and water. From this, it is reasonably expressed as,
in which ∆GDDAA means the Gibbs free energy of tetrahedral hydrogen bonding, RInterfacial water/volume is the molecular number ratio of interfacial water layer to volume, and nHB means the average hydrogen bonding number per molecule. For tetrahedral hydrogen bonding, nHB is equal to 2.
3. Hydrophobic Effects
When a solute is embedded in liquid water, it is thermodynamically equivalent to form the solute-water interface. After the solute is treated as a sphere, the RInterfacial water/volume is 4·rH2O/R, in which R means the radius of solute. The hydration free energy is the free energy associated with the transfer of solute from vacuum to water. Therefore, as the sphere solute is dissolved into water, hydration free energy may be described as (Figure 7),
in which ΔGWater-water is the Gibbs free energy of water, rH2O is the average radius of a H2O molecule. For water at 293 K and 0.1 MPa, Gibbs free energy (ΔGWater-water) is −1500 cal/mol [78]. Additionally, the average volume of a water molecule is 3 × 10−29 m3 at ambient conditions. After the water molecule is regarded as a sphere, the corresponding diameter is 3.8 Å, and rH2O is 1.9 Å. In addition, the Gibbs free energy of tetrahedral hydrogen bonding (ΔGDDAA) is calculated to be −2.66 kJ/mol at 293 K and 0.1 MPa.
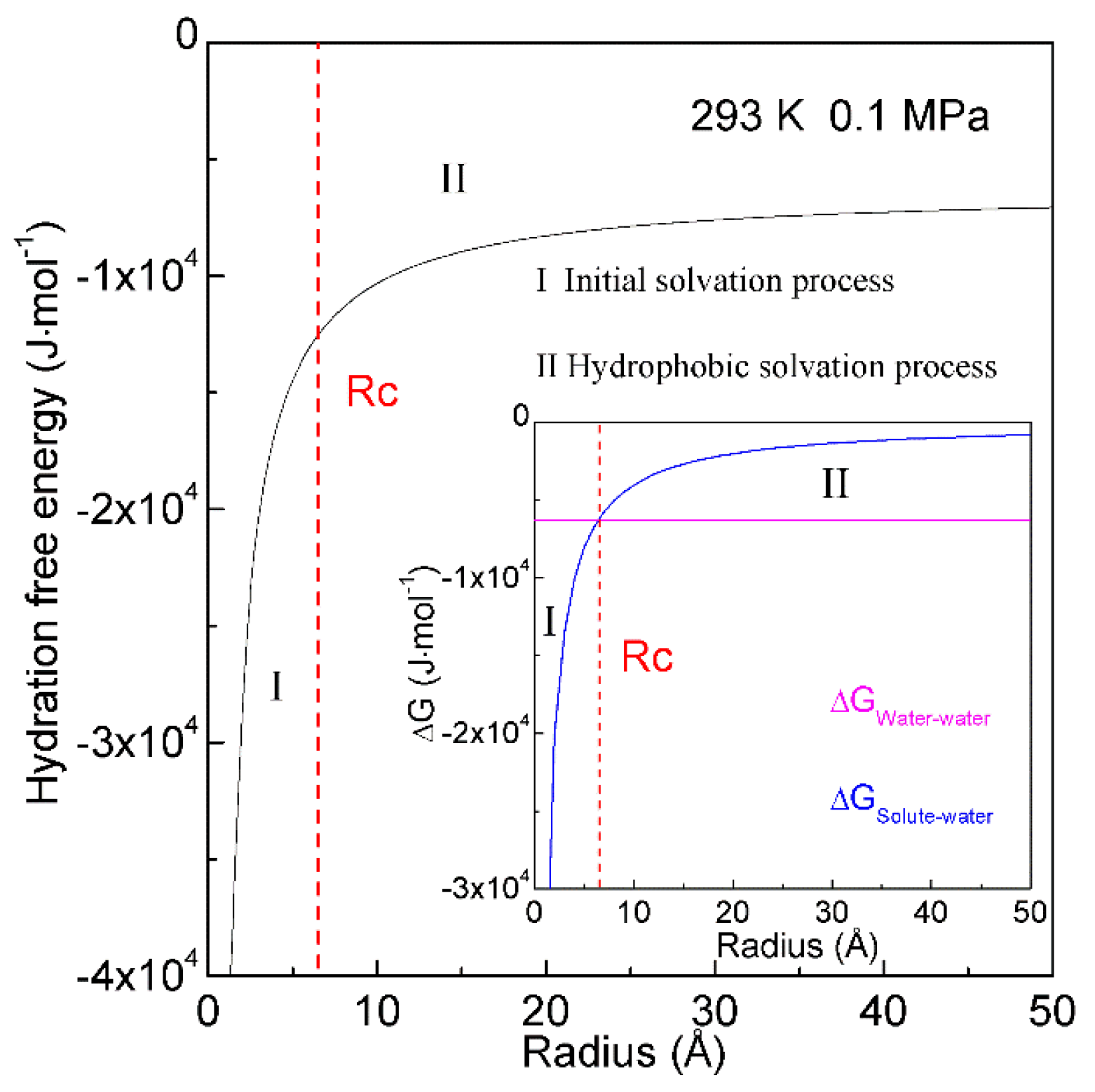
Figure 7. Hydration free energy at 293 K and 0.1 MPa. Hydration free energy is related to the size of solute, and critical radius (Rc) is expected. With increasing the solute size, it is divided into initial and hydrophobic solvation processes.
In thermodynamics, the lower the hydration free energy, the more stable the system is. Because hydration free energy is the sum of ∆GWater-water and ∆GSolute-water, it may be dominated by the Gibbs energy of bulk water (∆GWater-water) or interfacial water (∆GSolute-water). Of course, it is related to the size of dissolved solute. Therefore, the structural transition may be expected to occur as ∆GWater-water being equal to ∆GSolute-water,
in which Rc means the critical radius of dissolved solute [34].
With increasing the solute size, it is reasonably divided into the initial (ΔGWater-water < ΔGSolute-water) and hydrophobic (ΔGWater-water > ΔGSolute-water) solvation processes. The Gibbs free energy between solute and water (ΔGSolute-water) is inversely proportional to the solute size (1/R), which is related to the ratio of surface area to volume. Therefore, various dissolved behaviors of solutes in aqueous solutions may be expected in initial and hydrophobic processes, which may be related to the solute size (or concentrations) (Figure 8).
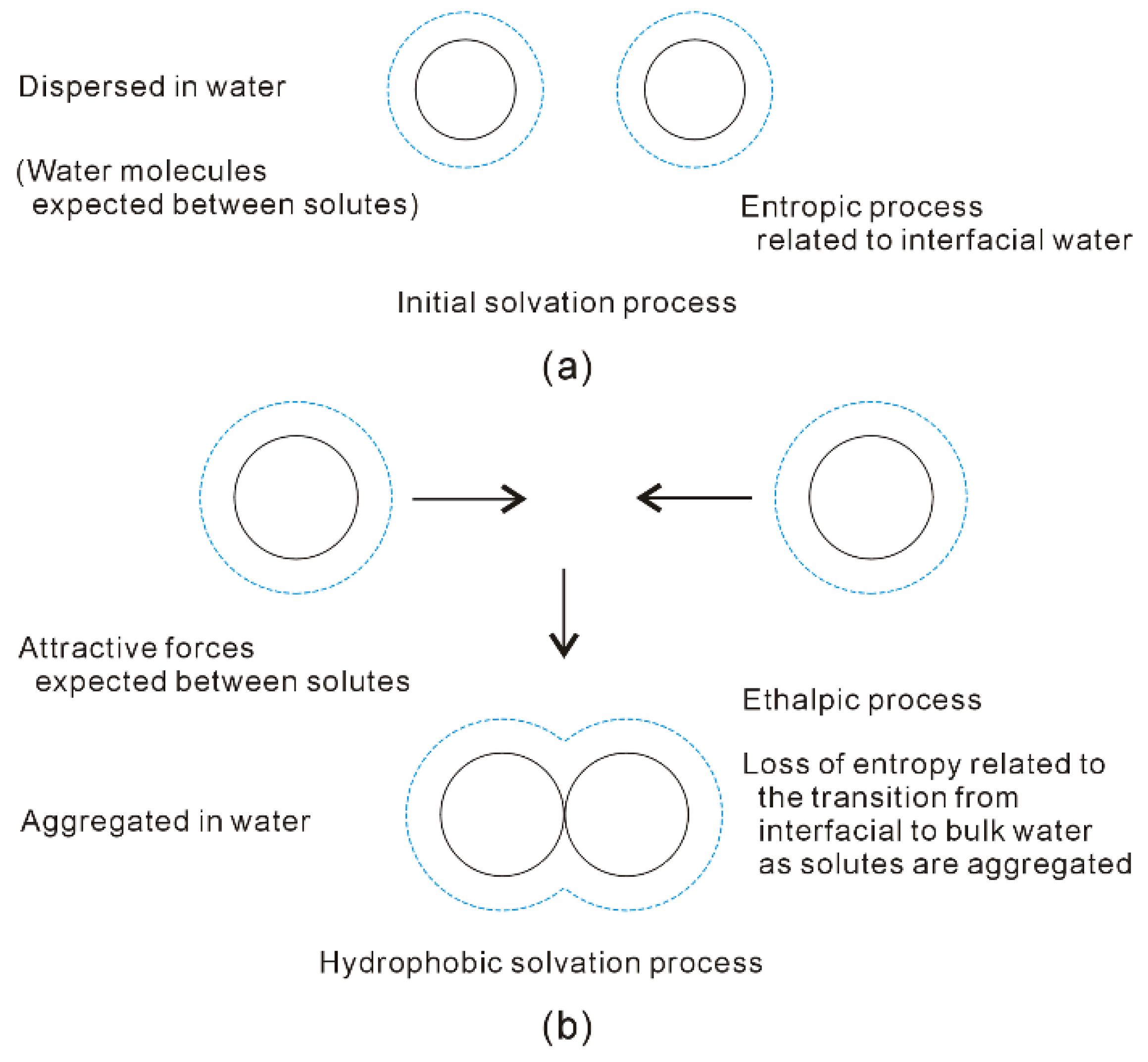
Figure 8. Different dissolved behaviors of solutes in aqueous solutions may be expected in initial (a) and hydrophobic (b) solvation processes.
In the initial solvation process, ΔGSolute-water is lower than ΔGWater-water (both of them are negative), or the solute size is smaller than Rc. Therefore, hydration free energy is dominated by the Gibbs free energy of interfacial water (ΔGSolute-water) [34][36]. To become more thermodynamically stable, this is fulfilled through maximizing the |ΔGSolute-water|. In other words, it is achieved through maximizing the ratio of surface area to volume of dissolved solutes. Therefore, the solutes may be dispersed in aqueous solutions, and water molecules are found between them (Figure 8). In addition, hydration free energy is proportional to the volume (or concentrations) of dissolved solutes.
Additionally, the dissolved solute mainly affects the hydrogen-bonded networks of interfacial water, DA hydrogen bondings tend to form within interfacial water layer [34]. In the initial solvation process, hydration free energy may be related to DA hydrogen bondings. In comparison with DDAA (tetrahedral) structural motif, DA hydrogen bonding owns weaker hydrogen bonding energy, and higher entropy. Therefore, the driving force may be thermodynamically ascribed to the increase of entropy arising from interfacial water [34].
In hydrophobic solvation process, Gibbs free energy of interfacial water is higher than bulk water (ΔGSolute-water > ΔGWater-water). To be more thermodynamically stable, this may be fulfilled through maximizing |ΔGWater-water|. Indeed, this is accompanied with the minimization of Gibbs free energy of interfacial water (|ΔGSolute-water|). From the above, the ΔGSolute-water is related to the ratio of surface area to volume of solutes. It can be derived that the dissolved solutes may be aggregated in solutions in order to maximize the hydrogen bondings of water [34][36]. Therefore, the “attractive” forces may be expected between solutes in hydrophobic solvation process (Figure 8).
Due to the existence of DDAA (tetrahedral) hydrogen bondings in bulk water, this leads to ΔGWater-water being lower than ΔGSolute-water. In comparison with DA hydrogen bonding, tetrahedral hydrogen-bonded networks own the stronger hydrogen bonding energy, and lower entropy [32]. Regarding the “attractive” force between solutes, it may be ascribed to be an enthalpic process, which is related to DDAA (tetrahedral) hydrogen bondings in bulk water. As the solutes are aggregated in water, they are also accompanied with the loss of entropy, which is related to the transition from interfacial to bulk water (Figure 8). Additionally, hydration free energy is proportional to the surface area of dissolved solutes.
To investigate the thermodynamic properties of hydrophobic interactions, the PMFs can be determined through MD simulations on C60-C60 fullerenes in water, and CH4-CH4 in water at different temperature (300 K, 320 K and 340 K) (Unpublished data). Based on the calculated ΔGWater-induced for C60-C60 in water, and CH4-CH4 in water, these can be applied to determine the thermodynamic functions as the solutes are associated in water (Figure 9). To be more thermodynamically stable, the two CH4 molecules are engaged in the solvent-separated conformation. In thermodynamics, it is driven by the entropy contributions related to interfacial water (Figure 9). However, the two fullerenes tend to be accumulated in solutions. Thermodynamically, it is dominated by enthalpy related to maximizing the hydrogen bondings of water. Additionally, this is also accompanied with the loss of entropy. Indeed, this is related to the transition from interfacial to bulk water as the fullerenes are aggregated in solutions, especially as the distance between them is less than 13 Å (Figure 9). Therefore, various thermodynamic driving forces may be expected in initial and hydrophobic solvation processes, which may be also in agreement with the above structural studies.
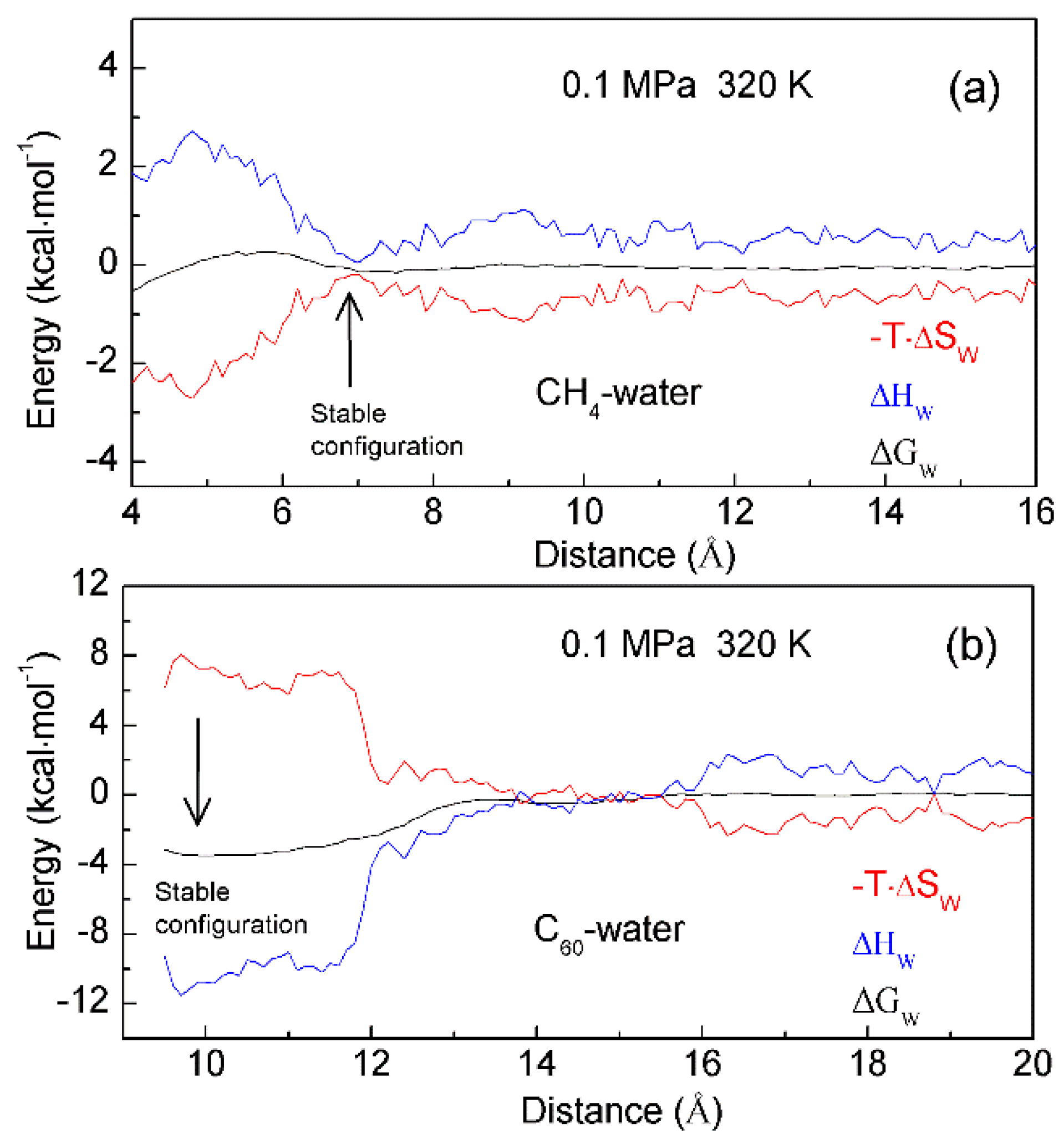
Figure 9. The thermodynamic characteristics of hydrophobic interactions. Based on the calculated PMFs of C60-C60 fullerenes and CH4-CH4 in water at different temperatures (300 K, 320 K and 340 K), water contributions to Gibbs energy (ΔGW) are determined, which are used to calculate the enthalpic (ΔHW) and entropic (-T·ΔSW) contributions. Different thermodynamic characteristics may be expected in initial (a) and hydrophobic (b) solvation processes.
In addition, enthalpy-entropy compensation (EEC) has been attracting attention because it is involved in many research fields [79][80][81][82][83], especially for the understanding of molecular recognition and drug design. In thermodynamics, EEC means that, if, for the particular reaction, ΔH and ΔS are changing in one direction (either increasing or decreasing), their changes that are transformed into ΔG are mutually compensated, and there is little change in the value of ΔG. To understand the nature of EEC, many works have been carried out. This means that EEC is real, very common, and a consequence of the properties of liquid water. Based on the calculated thermodynamics functions (Figure 9), hydrophobic interactions are closely related to EEC. Therefore, water plays a vital role in the process of EEC. Of course, this has been demonstrated in the work of Gilli et al. [83].
The dissolved solutes mainly affect the structure of interfacial water. Therefore, the effects of solutes on water structure may be related to the surfaces of solutes to be available for interfacial water. Owing to hydrophobic interactions, the dissolved solutes are attracted and tend to be aggregated in aqueous solutions in order to maximize the hydrogen bondings of water. In fact, the solutes coming into contact undoubtedly leads to the decrease of the solute surfaces available for interfacial water. Therefore, the Gibbs free energy of interfacial water may be described as,
where γ is named as the geometric factor [36]. It is proposed to reflect the changes of solute surfaces while solutes are accumulated in water. Naturally, the solutes are rarely rigid. As they are dissolved in solutions, this may be accompanied by the changes of solute volume. From this, γ may be generally expressed as,
in which rSeparation means the distance between solutes. When the solutes come into contact, the corresponding distance between them is termed ‘the hydrophobic radius’ (RH) [36]. As the solutes are aggregated in water, it may be divided into H1w and H2s hydrophobic solvation processes, respectively.
Water molecules may be found between the dissolved solutes in H1w hydrophobic process, the separation between solutes is larger than RH (>RH), or γ is 1 [36]. Due to hydrophobic interactions, the solutes are attracted to approaching each other. This decreases the distance between the solutes, and the water molecules in the region between them are expelled into bulk water. Therefore, hydrophobic interactions are fulfilled by the rearrangement of water molecules. Additionally, energy barriers may be expected in the H1w process, due to the expelled water molecules. Thermodynamically, the dissolved solutes are expected to approach each other in the direction with the lowest energy barrier, in which less water molecules may be expelled. From the above, the directional nature may be expected in the H1w hydrophobic process [37].
References
- Scheraga, H.A. Theory of hydrophobic interactions. J. Biomol. Struct. Dyn. 1998, 16, 447–460.
- Blokzijl, W.; Engberts, J.B.F.N. Hydrophobic effects. Opinions and facts. Angew. Chem. Int. Ed. 1993, 32, 1545–1579.
- Makowski, M.; Czaplewski, C.; Liwo, A.; Scheraga, H.A. Potential of mean force of association of large hydrophobic particles: Toward the nanoscale limit. J. Phys. Chem. B 2010, 114, 993–1003.
- Sobolewski, E.; Makowski, M.; Czaplewski, C.; Liwo, A.; Ołdziej, S.; Scheraga, H.A. Potential of mean force of hydrophobic association: Dependence on solute size. J. Phys. Chem. B 2007, 111, 10765–10774.
- Bartosik, A.; Wiśniewska, M.; Makowski, M. Potentials of mean force for hydrophobic interactions between hydrocarbons in water solution: Dependence on temperature, solute shape, and solute size. J. Phys. Org. Chem. 2015, 28, 10–16.
- Hansch, C.; Fujita, T. p-σ-π analysis. A method for the correlation of biological activity and chemical structure. J. Am. Chem. Soc. 1964, 86, 1616–1626.
- Kujawski, J.; Popielarska, H.; Myka, A.; Drabińska, B.; Bernard, M.K. The log P parameter as a molecular descriptor in the computer-aided drug design-an overview. Comput. Meth. Sci. Technol. 2012, 18, 81–88.
- Frank, H.S.; Evans, M.W. Free volume and entropy in condensed systems III. Entropy in binary liquid mixtures; partial molal entropy in dilute solutions; structure and thermodynamics in aqueous electrolytes. J. Chem. Phys. 1945, 13, 507–532.
- Davis, J.G.; Gierszal, K.P.; Wang, P.; Ben-Amotz, D. Water structural transformation at molecular hydrophobic interfaces. Nature 2012, 491, 582–585.
- Galamba, N. Water’s structure around hydrophobic solutes and the iceberg model. J. Phys. Chem. B 2013, 117, 2153–2159.
- Raschke, T.M.; Levitt, M. Nonpolar solutes enhance water structure within hydration shells while reducing interactions between them. Proc. Natl. Acad. Sci. USA 2005, 102, 6777–6782.
- Rezus, Y.L.A.; Bakker, H.J. Observation of immobilized water molecules around hydrophobic groups. Phys. Rev. Lett. 2007, 99, 148301.
- Turner, J.; Soper, A.K.; Finney, J.L. A neutron-diffraction study of tetramethylammonium chloride in aqueous solution. Mol. Phys. 1990, 70, 679–700.
- Qvist, J.; Halle, B. Thermal signature of hydrophobic hydration dynamics. J. Am. Chem. Soc. 2008, 130, 10345–10353.
- Buchanan, P.; Aldiwan, N.; Soper, A.K.; Creek, J.L.; Koh, C.A. Decreased structure on dissolving methane in water. Chem. Phys. Lett. 2005, 415, 89–93.
- Bakulin, A.A.; Liang, C.; Jansen, T.L.; Wiersma, D.A.; Bakker, H.J.; Pshenichnikov, M.S. Hydrophobic solvation: A 2D IR spectroscopic inquest. Acc. Chem. Res. 2009, 42, 1229–1238.
- Sun, Q. The effects of dissolved hydrophobic and hydrophilic groups on water structure. J. Solution Chem. 2020, 49, 1473–1484.
- Kauzmann, W. Some factors in the interpretation of protein denaturation. Adv. Protein Chem. 1959, 14, 1–63.
- Ferguson, S.B.; Seward, E.M.; Diederich, F.; Sanford, E.M.; Chou, A.; Inocencio-Szweda, P.; Knobler, C.B. Strong enthalpically driven complexation of neutral benzene guests in aqueous solution. J. Org. Chem. 1988, 53, 5593–5595.
- Baron, R.; Setny, P.; McCammon, J.A. Water in cavity-ligand recognition. J. Am. Chem. Soc. 2010, 132, 12091–12097.
- Setny, P.; Baron, R.; McCammon, J.A. How can hydrophobic association be enthalpy driven? J. Chem. Theory Comput. 2010, 6, 2866–2871.
- Hummer, G. Molecular binding under water’s influence. Nat. Chem. 2010, 2, 906–907.
- Huang, D.M.; Geissler, P.L.; Chandler, D. Scaling of hydrophobic solvation free energies. J. Phys. Chem. B 2001, 105, 6704–6709.
- Lum, K.; Chandler, D.; Weeks, J.D. Hydrophobicity at small and large length scales. J. Phys. Chem. B 1999, 103, 4570–4577.
- Chandler, D. Interfaces and the driving force of hydrophobic assembly. Nature 2005, 437, 640–647.
- Huang, D.M.; Chandler, D. Temperature and length scale dependence of hydrophobic effects and their possible implications for protein folding. Proc. Natl. Acad. Sci. USA 2000, 97, 8324–8327.
- Ball, P. Water as an active constituent in cell biology. Chem. Rev. 2008, 108, 74–108.
- Ball, P. Water is an active matrix of life for cell and molecular biology. Proc. Natl. Acad. Sci. USA 2017, 114, 13327–13335.
- Ball, P. More than a bystander. Nature 2011, 478, 467–468.
- Sun, Q. The Raman OH stretching bands of liquid water. Vib. Spectrosc. 2009, 51, 213–217.
- Sun, Q. Raman spectroscopic study of the effects of dissolved NaCl on water structure. Vib. Spectrosc. 2012, 62, 110–114.
- Sun, Q. Local statistical interpretation for water structure. Chem. Phys. Lett. 2013, 568, 90–94.
- Sun, Q.; Guo, Y. Vibrational sum frequency generation spectroscopy of the air/water interface. J. Mol. Liq. 2016, 213, 28–32.
- Sun, Q. The physical origin of hydrophobic effects. Chem. Phys. Lett. 2017, 672, 21–25.
- Sun, Q.; Zhang, M.X.; Cui, S. The structural origin of hydration repulsive force. Chem. Phys. Lett. 2019, 714, 30–36.
- Sun, Q.; Su, X.W.; Cheng, C.B. The dependence of hydrophobic interactions on the solute size. Chem. Phys. 2019, 516, 199–205.
- Sun, Q.; Wang, W.Q.; Cui, S. Directional nature of hydrophobic interactions: Implications for the mechanism of molecular recognition. Chem. Phys. 2021, 547, 111200.
- Sun, Q.; Cui, S.; Zhang, M.X. Homogeneous nucleation mechanism of NaCl in aqueous solutions. Crystals 2020, 10, 107.
- Stanley, H.E.; Teixeira, J. Interpretation of the unusual behavior of H2O and D2O at low temperatures: Tests of a percolation model. J. Chem. Phys. 1980, 73, 3404–3422.
- Nilsson, A.; Pettersson, L.G.M. Perspective on the structure of liquid water. Chem. Phys. 2011, 389, 1–34.
- Röntgen, W.C. Ueber die Constitution des flüssigen Wassers. Ann. Phys. 1892, 281, 91–97.
- Russo, J.; Tanaka, H. Understanding water’s anomalies with locally favoured structures. Nat. Commun. 2014, 5, 3556.
- Hamm, P. Markov state model of the two-state behaviour of water. J. Chem. Phys. 2016, 145, 134501.
- Shi, R.; Tanaka, H. Microscopic structural descriptor of liquid water. J. Chem. Phys. 2018, 148, 124503.
- Skinner, L.B.; Huang, C.; Schlesinger, D.; Pettersson, L.G.M.; Nilsson, A.; Benmore, C.J. Benchmark oxygen-oxygen pair-distribution function of ambient water from X-ray diffraction measurements with a wide Q-range. J. Chem. Phys. 2013, 138, 074506.
- Hura, G.; Sorenson, J.M.; Glaeser, R.M.; Head-Gordon, T. A high-quality X-ray scattering experiment on liquid water at ambient conditions. J. Chem. Phys. 2000, 113, 9140.
- Misquitta, A.J.; Szalewicz, K. Intermolecular forces from asymptotically corrected density functional description of monomers. Chem. Phys. Lett. 2002, 357, 301–306.
- Misquitta, A.J.; Jeziorski, B.; Szalewicz, K. Dispersion energy from density-functional theory description of monomers. Phys. Rev. Lett. 2003, 91, 033201.
- Hoja, J.; Sax, A.F.; Szalewicz, K. Is electrostatics sufficient to describe hydrogen-bonding interactions? Chem. Eur. J. 2014, 20, 2292–2300.
- Fraley, P.E.; Rao, K.N. High resolution infrared spectra of water vapor: ν1 and ν3 band of H216O. J. Mol. Spectrosc. 1969, 29, 348–364.
- Ludwig, R. The effect of hydrogen bonding on the thermodynamic and spectroscopic properties of molecular clusters and liquids. Phys. Chem. Chem. Phys. 2002, 4, 5481–5487.
- Smith, J.D.; Cappa, C.D.; Wilson, K.R.; Cohen, R.C.; Geissler, P.L.; Saykally, R.J. Unified description of temperature-dependent hydrogen-bond rearrangements in liquid water. Proc. Natl. Acad. Sci. USA 2005, 102, 14171–14174.
- Huang, C.; Wikfeldt, K.T.; Tokushima, T.; Nordlund, D.; Harada, Y.; Bergmann, U.; Niebuhr, M.; Weiss, T.M.; Horikawa, Y.; Leetmaa, M.; et al. The inhomogeneous structure of water at ambient conditions. Proc. Natl. Acad. Sci. USA 2009, 106, 15214–15218.
- Kim, K.H.; Späh, A.; Pathak, H.; Perakis, F.; Mariedahl, D.; Amann-Winkel, K.; Sellberg, J.A.; Lee, J.H.; Kim, S.; Park, J.; et al. Maxima in the thermodynamic response and correlation functions of deeply supercooled water. Science 2017, 358, 1589–1593.
- Matsumoto, M.; Saito, S.; Ohmine, I. Molecular dynamics simulation of the ice nucleation and growth process leading to water freezing. Nature 2002, 416, 409–413.
- Moore, E.B.; Molinero, V. Structural transformation in supercooled water controls the crystallization rate of ice. Nature 2011, 479, 506–508.
- Fitzner, M.; Sosso, G.C.; Cox, S.J.; Michaelides, A. Ice is born in low-mobility regions of supercooled liquid water. Proc. Natl. Acad. Sci. USA 2019, 116, 2009–2014.
- Trudu, F.; Donadio, D.; Parrinello, M. Freezing of a Lennard-Jones fluid: From nucleation to spinodal regime. Phys. Rev. Lett. 2006, 97, 105701.
- Berryman, J.T.; Anwar, M.; Dorosz, S.; Schilling, T. The early crystal nucleation process in hard spheres shows synchronised ordering and densification. J. Chem. Phys. 2016, 145, 211901.
- Desgranges, C.; Delhommelle, J. Can ordered precursors promote the nucleation of solid solutions? Phys. Rev. Lett. 2019, 123, 195701.
- Speedy, R.J. Limiting forms of the thermodynamic divergences at the conjectured stability limits in superheated and supercooled water. J. Phys. Chem. 1982, 86, 3002–3005.
- Poole, P.H.; Sciortino, F.; Essmann, U.; Stanley, H.E. Phase behaviour of metastable water. Nature 1992, 360, 324–328.
- Sastry, S.; Debenedetti, P.G.; Sciortino, F.; Stanley, H.E. Singularity-free interpretation of the thermodynamics of supercooled water. Phys. Rev. E 1996, 53, 6144.
- Angell, C.A. Insights into phases of liquid water from study of its unusual glass-forming properties. Science 2008, 319, 582–587.
- Kim, K.H.; Amann-Winkel, K.; Giovambattista, N.; Späh, A.; Perakis, F.; Pathak, H.; Parada, M.L.; Yang, C.; Mariedahl, D.; Eklund, T.; et al. Experimental observation of the liquid-liquid transition in bulk supercooled water under pressure. Science 2020, 370, 978–982.
- Palmer, J.C.; Martelli, F.; Liu, Y.; Car, R.; Panagiotopoulos, A.Z.; Debenedetti, P.G. Metastable liquid-liquid transition in a molecular model of water. Nature 2014, 510, 385–388.
- Debenedetti, P.G.; Sciortino, F.; Zerze, G.H. Second critical point in two realistic models of water. Science 2020, 369, 289–292.
- Gartner, T.E.; Zhang, L.; Piaggi, P.M.; Car, R.; Panagiotopoulos, A.Z.; Debenedetti, P.G. Signatures of a liquid-liquid transition in an ab initio deep neural network model for water. Proc. Natl. Acad. Sci. USA 2020, 117, 26040–26046.
- Handle, P.H.; Loerting, T.; Sciortino, F. Supercooled and glassy water: Metastable liquid(s), amorphous solid(s), and a no-man’s land. Proc. Natl. Acad. Sci. USA 2017, 114, 13336–13344.
- Collins, K.D.; Neilson, G.W.; Enderby, J.E. Ions in water: Characterizing the forces that control chemical processes and biological structure. Biophys. Chem. 2007, 128, 95–104.
- Cappa, C.D.; Smith, J.D.; Messer, B.M.; Cohen, R.C.; Saykally, R.J. Effects of cations on the hydrogen bond network of liquid water: New results from X-ray absorption spectroscopy of liquid microjets. J. Phys. Chem. B 2006, 110, 5301–5309.
- Omta, A.W.; Kropman, M.F.; Woutersen, S.; Bakker, H.J. Negligible effect of ions on the hydrogen-bond structure in liquid water. Science 2003, 301, 347–349.
- Moilanen, D.E.; Wong, D.; Rosenfeld, D.E.; Fenn, E.E.; Fayer, M.D. Ion-water hydrogen-bond switching observed with 2D IR vibrational echo chemical exchange spectroscopy. Proc. Natl. Acad. Sci. USA 2009, 106, 375–380.
- Turton, D.A.; Hunger, J.; Hefter, G.; Buchner, R.; Wynne, K. Glasslike behavior in aqueous electrolyte solutions. J. Chem. Phys. 2008, 128, 161102.
- Thompson, H.; Soper, A.K.; Ricci, M.A.; Bruni, F.; Skipper, N.T. The three-dimensional structure of water confined in nanoporous vycor glass. J. Phys. Chem. B 2007, 111, 5610–5620.
- Giri, A.K.; Teixeira, F.; Cordeiro, M.N.D.S. Structure and kinetics of water in highly confined conditions: A molecular dynamics simulation study. J. Mol. Liq. 2018, 268, 625–636.
- Winarto; Takaiwa, D.; Yamamoto, E.; Yasuoka, K. Structures of water molecules in carbon nanotubes under electric fields. J. Chem. Phys. 2015, 142, 124701.
- Dorsey, N.E. Properties of Ordinary Water Substance; ACS Monograph No. 81; Reinhold Publishing Corp.: New York, NY, USA, 1940.
- Chodera, J.D.; Mobley, D.L. Entropy-enthalpy compensation: Role and ramifications in biomolecular ligand recognition and design. Annu. Rev. Biophys. 2013, 42, 121–142.
- Dragan, A.I.; Read, C.M.; Crane-Robinson, C. Enthalpy-entropy compensation: The role of solvation. Eur. Biophys. J. 2017, 46, 301–308.
- Breiten, B.; Lockett, M.R.; Sherman, W.; Fujita, S.; Al-Sayah, M.; Lange, H.; Bowers, C.M.; Heroux, A.; Krilov, G.; Whitesides, G.M. Water networks contribute to enthalpy/entropy compensation in protein-ligand binding. J. Am. Chem. Soc. 2013, 135, 15579–15584.
- Sharp, K. Entropy-enthalpy compensation: Fact or artifact? Protein Sci. 2001, 10, 661–667.
- Gilli, P.; Ferrett, V.; Gilli, G.; Borea, P.A. Enthalpy-entropy compensation in drug-receptor binding. J. Phys. Chem. 1994, 98, 1515–1518.
More
Information
Subjects:
Chemistry, Physical
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.5K
Revisions:
5 times
(View History)
Update Date:
01 Nov 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No