Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 3 by Conner Chen and Version 5 by Conner Chen.
Hydrophobic interactions are involved in and believed to be the fundamental driving force of many chemical and biological phenomena in aqueous environments.
- water
- solute
- interface
- hydrogen bonding
- hydrophobic effects
1. Introduction
Hydrophobic effects refer to the tendency of nonpolar molecules (or parts of molecules) to be aggregated in water. They are involved in and believed to be the fundamental driving force in many chemical and biological phenomena in aqueous solutions, such as molecular recognition, protein folding, formation and stability of micelles, biological membranes and macromolecular complexes, surfactant aggregation, coagulation, complexation, detergency, and the formation of gas clathrates [1][2][3][4][5]. To date, numerous experimental and theoretical works have been carried out to understand the physical origin of hydrophobic effects.
In general, hydrophobicity is expressed through the empirically calculated logarithm of partition coefficient (logP), which is widely used in drug design and medicinal chemistry [6][7]. Historically, the concept of hydrophobicity arose in the context of the low solubility of non-polar solutes in water. Experimentally, it has been found that the entropy change upon transferring a hydrocarbon from a nonpolar environment into water is large and negative. In 1945, Frank and Evans [8] suggested that the observed loss of entropy was related to the structural changes of liquid water as hydrophobic solutes were dissolved into water. They proposed that a kind of “cage”, consisting of water molecules, was formed around the solute. This ordered water structure, similar to the gas clathrate, was known as the “iceberg” model [8]. Since then, many works [9][10][11][12][13][14][15][16][17] are conducted to measure the water structural changes around hydrophobic surfaces. In accordance with the “iceberg” model, some studies [9][10][11][12] support the existence of increased tetrahedral order around small hydrophobic groups in aqueous solutions. However, the decrease of water structure around hydrophobic groups is also found in many works [13][14][15][16][17]. It is well known that the neutron scattering may be sensitive to the structure of water. From the neutron scattering experimental measurements on aqueous solutions containing tetramethylammonium chloride [13] and methane molecules [15], these do not suggest that water around these hydrophobic solutes may be more ordered than bulk water. To date, there remain strong debates on the “iceberg” structural model.
In 1959, based on the “iceberg” model, Kauzmann [18] introduced the concept of hydrophobic interactions. When two “caged” hydrophobes come together, the “structured” water between solutes may be released into the bulk (Figure 1), which undoubtedly leads to the increase of entropy. Subsequently, the attractive force between these particles may be related to the entropy increase. Therefore, hydrophobic interactions are classically regarded to be entropy driven. However, the “classic” hydrophobic effects may be in contrast with some works. In Diederich et al. [19] work, they found that complexation of benzene in a cyclophane host molecule was enthalpy driven at room temperature, which was also accompanied with a slightly negative entropy change. Additionally, in Baron, Setny, and McCammon works [20][21], molecular dynamics (MD) simulations are used to investigate the binding in a hydrophobic receptor-ligand system. It is found that the association between the non-polar ligand and binding pocket may be driven by enthalpy and opposed by entropy. The “non-classic” hydrophobic effects are ascribed to the release of weakly hydrogen-bonded water molecules into the more strongly hydrogen-bonded bulk water [22].
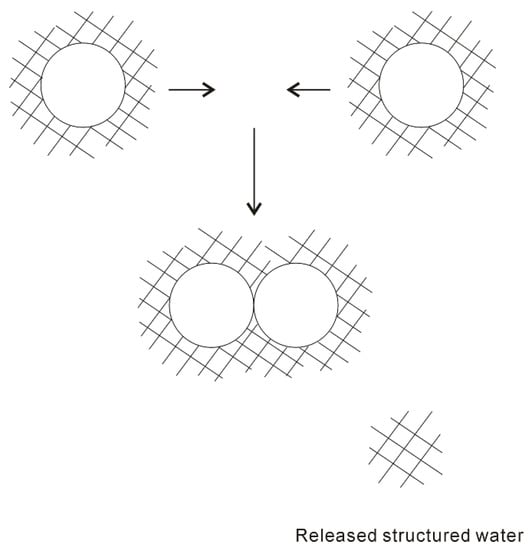
Figure 1. The “iceberg” structural model of hydrophobic effects. The ordered water structure is expected to form around the solute. As two “caged” solutes become together, the “structured” water in the region between them may be returned to the bulk.
In the “iceberg” structural model, water molecules can rearrange around a small hydrophobic solute, without losing their hydrogen bondings. However, as a large solute is embedded into water, hydrogen bondings of water may be broken at the surface of the solute, which may result in an enthalpic penalty. Therefore, hydrophobic interactions depend on the size of dissolved solute. In recent years, a theoretical approach was developed by Lum, Chandler, and Weeks (LCW) [23][24][25][26] to understand the dependence of hydrophobic interactions on solute size. Both the Gaussian density fluctuations related to small size and the physics of interfacial formation related to large size are incorporated in LCW theory [23][24][25][26]. It can be found that the hydration free energy grows linearly with the solvated volume for small solute, but grows linearly with the solvated surface area for large solute [25]. Therefore, the crossover may be expected between small and large regime, which takes place on the nanometer length scale [25].
Liquid water is generally believed to play a vital role in the process of hydrophobic interactions. In cell biology, water may be regarded as an active constituent, rather than a bystander [27][28][29]. According to some recent structural works [30][31][32][33][34][35][36][37][38] on liquid water and air-water interface, hydration free energy is determined. This is utilized to understand the nature of hydrophobic interactions. It is found that hydrophobic interactions may be related to the size of dissolved solute. With increasing the solute size, it is reasonably divided into initial and hydrophobic solvation processes [34]. Additionally, different dissolved behaviors of solutes are expected in initial and hydrophobic solvation processes, such as dispersed and accumulated distributions in aqueous environments. In addition, hydrophobic interactions may be ascribed to the structural competition between interfacial and bulk water [34].
2. Water Structure
Gibbs free energy (ΔG), related to the changes of enthalpy (ΔH) and entropy (ΔS), may be used to study whether a process is likely to take place. Thermodynamically, it is expressed as,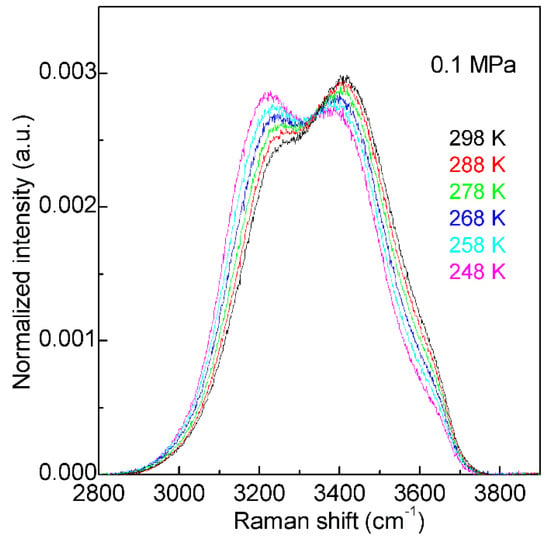
Figure 2. The Raman OH stretching bands of water from 298 to 248 K under 0.1 MPa. Based on normalized intensity, an isosbestic point is found around 3330 cm−1.
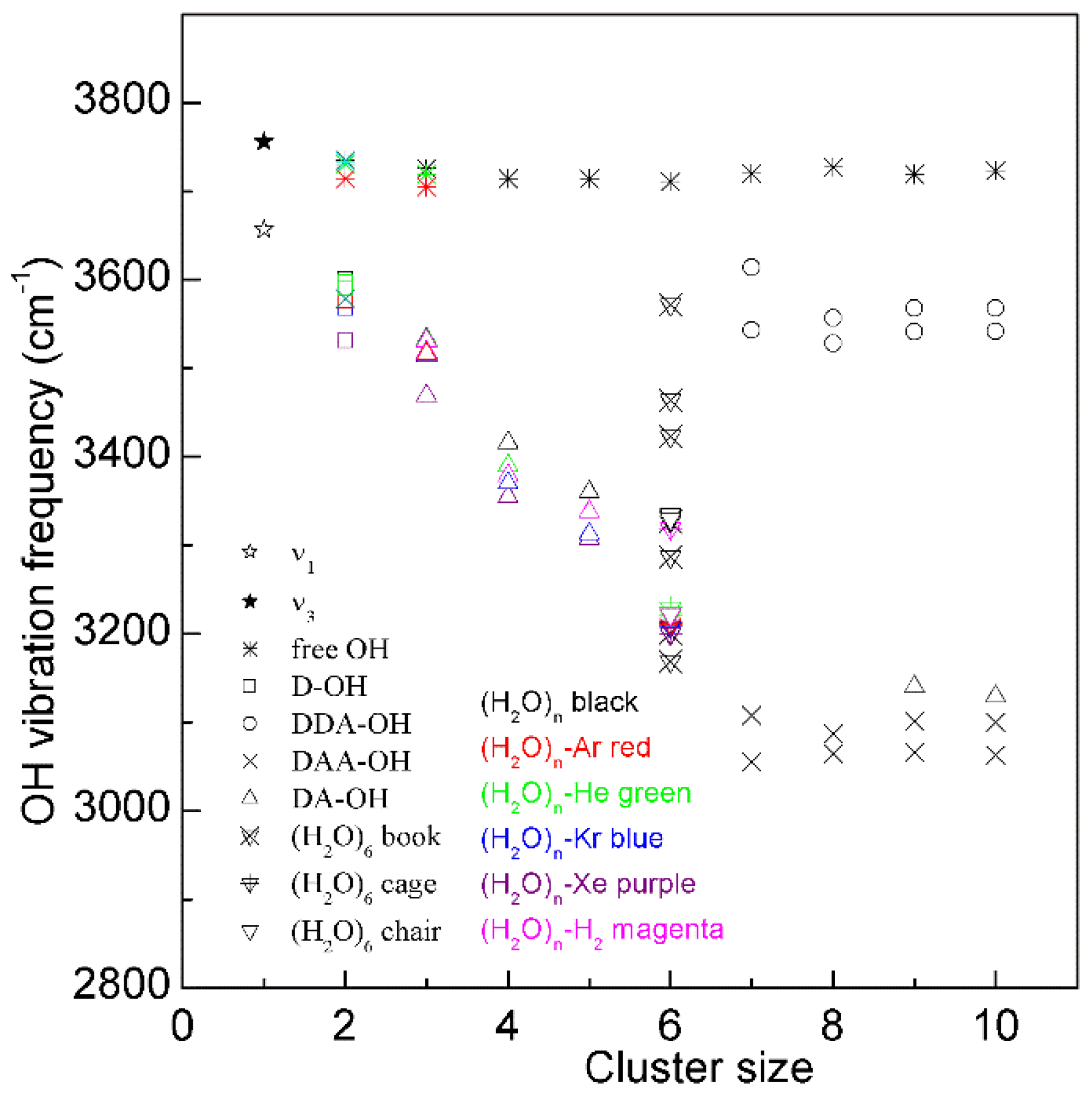
Figure 3. The dependence of the OH stretching frequency on hydrogen bondings of water molecular clusters, (H2O)n. Different symbols are used to discriminate OH vibrations engaged in various local hydrogen-bonded networks of a water molecule. Various structures of hexamers are also shown.
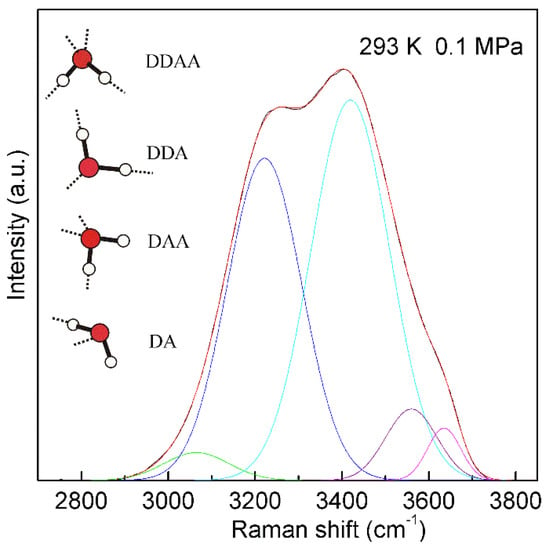
Figure 4. The Raman OH stretching band of ambient water may be deconvoluted into five sub-bands, located at 3041, 3220, 3430, 3572, and 3636 cm−1, and assigned to the νDAA-OH, νDDAA-OH, νDA-OH, νDDA-OH, and free OH symmetric stretching vibrations, respectively. At ambient conditions, the main local hydrogen-bonded networks for a water molecule are expected to be DDAA, DDA, DAA, and DA hydrogen bondings. Hydrogen bondings are drawn with dashed lines.
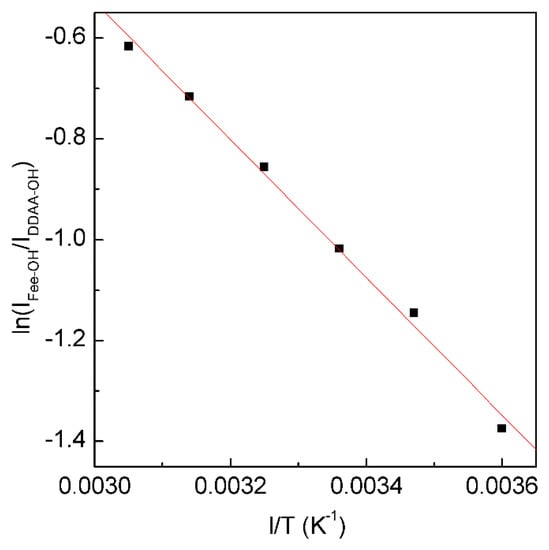
Figure 5. The dependence of ln(IFree-OH/IDDAA-OH) on 1/T. The solid line represents linear fit (R2 = 0.9975) with a slope of −∆H/R. This is used to determine the thermodynamic characteristics of tetrahedral hydrogen bonding.
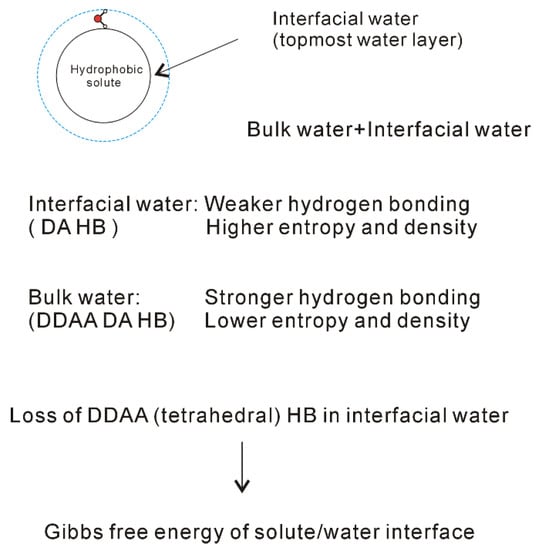
Figure 6. Structural changes across the solute-water interface. The dissolved solute mainly affects the structure of interfacial water (topmost water layer at the interface).
3. Hydrophobic Effects
When a solute is embedded in liquid water, it is thermodynamically equivalent to form the solute-water interface. After the solute is treated as a sphere, the RInterfacial water/volume is 4·rH2O/R, in which R means the radius of solute. The hydration free energy is the free energy associated with the transfer of solute from vacuum to water. Therefore, as the sphere solute is dissolved into water, hydration free energy may be described as (Figure 7),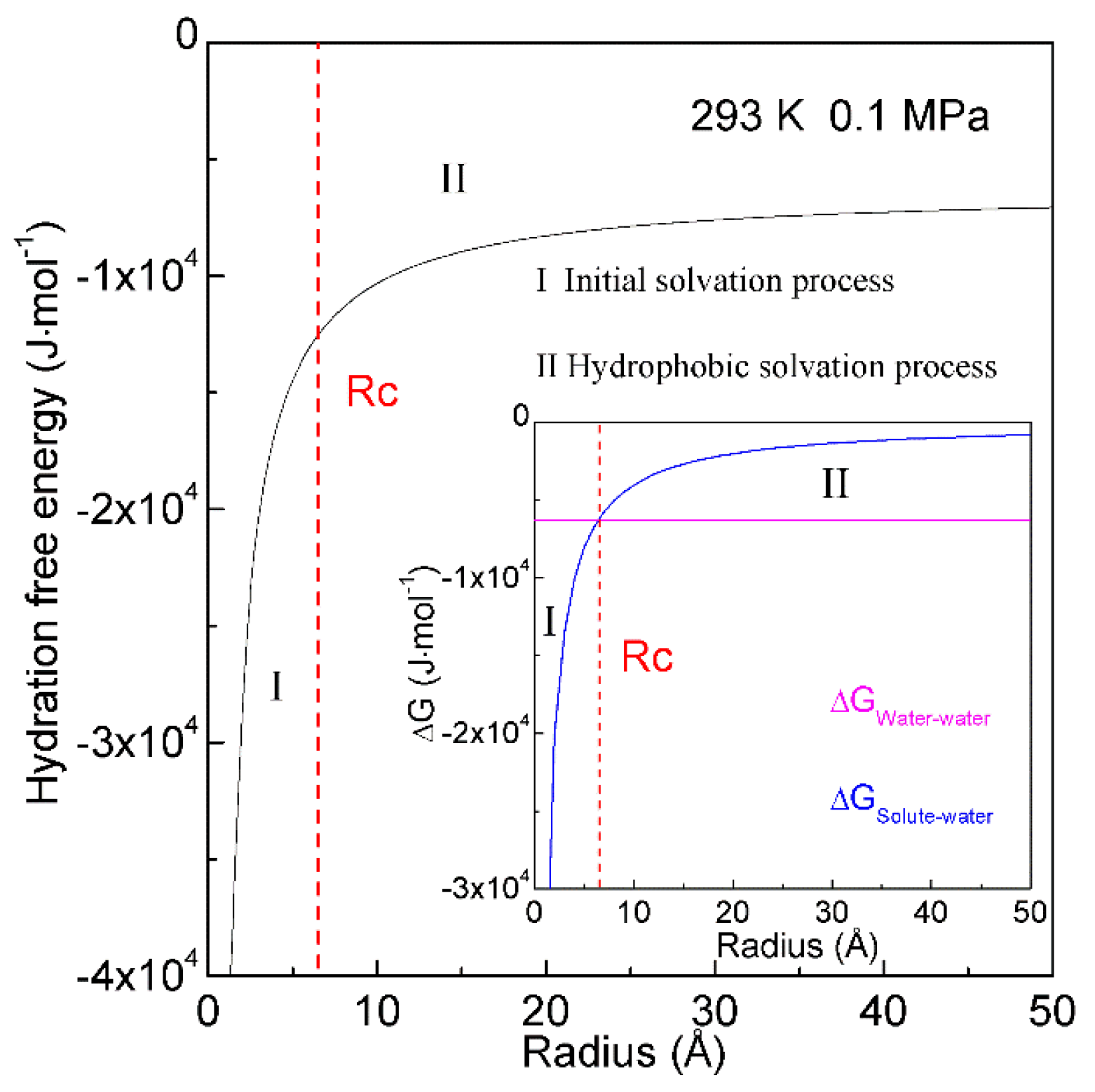
Figure 7. Hydration free energy at 293 K and 0.1 MPa. Hydration free energy is related to the size of solute, and critical radius (Rc) is expected. With increasing the solute size, it is divided into initial and hydrophobic solvation processes.
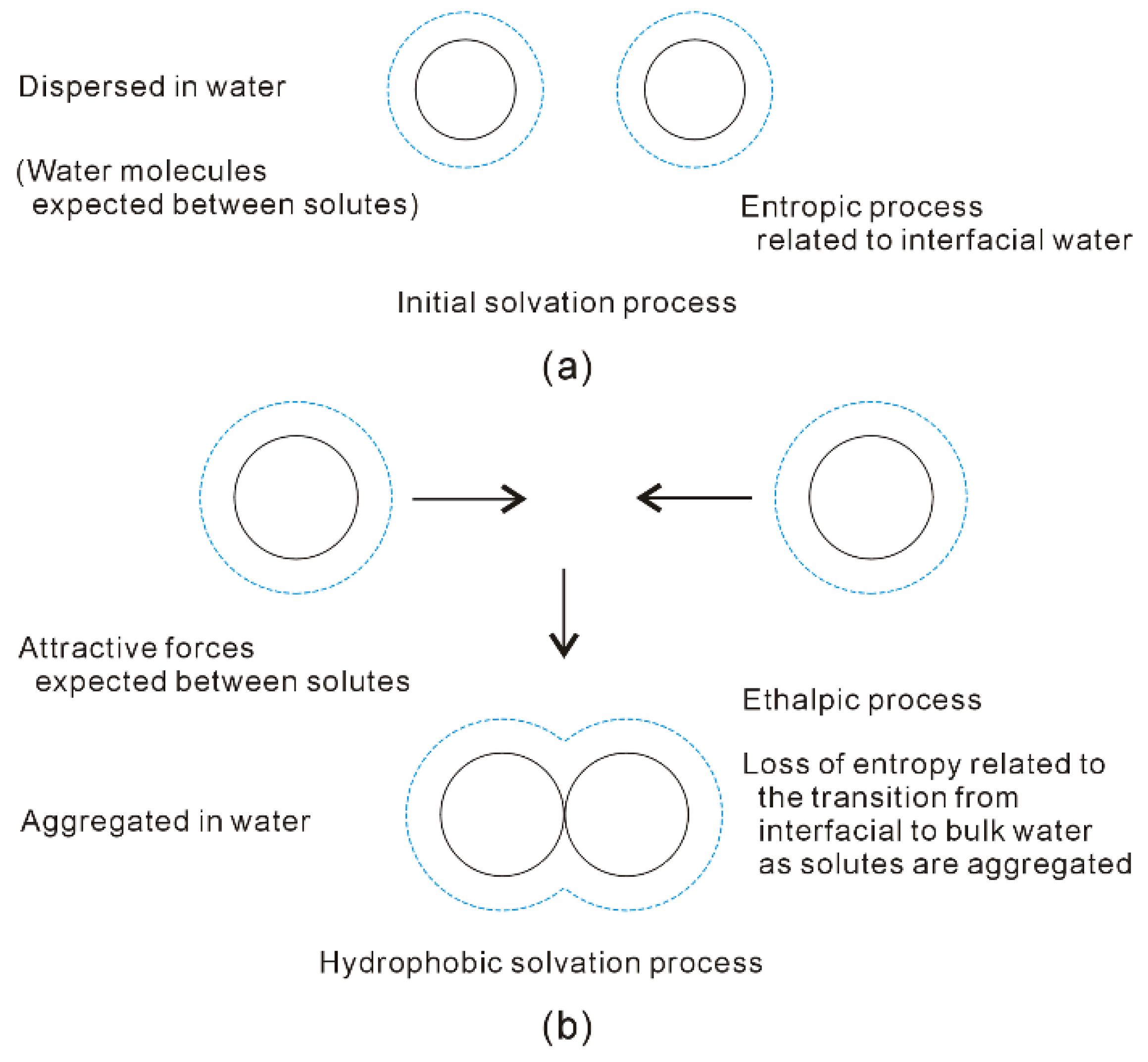
Figure 8. Different dissolved behaviors of solutes in aqueous solutions may be expected in initial (a) and hydrophobic (b) solvation processes.
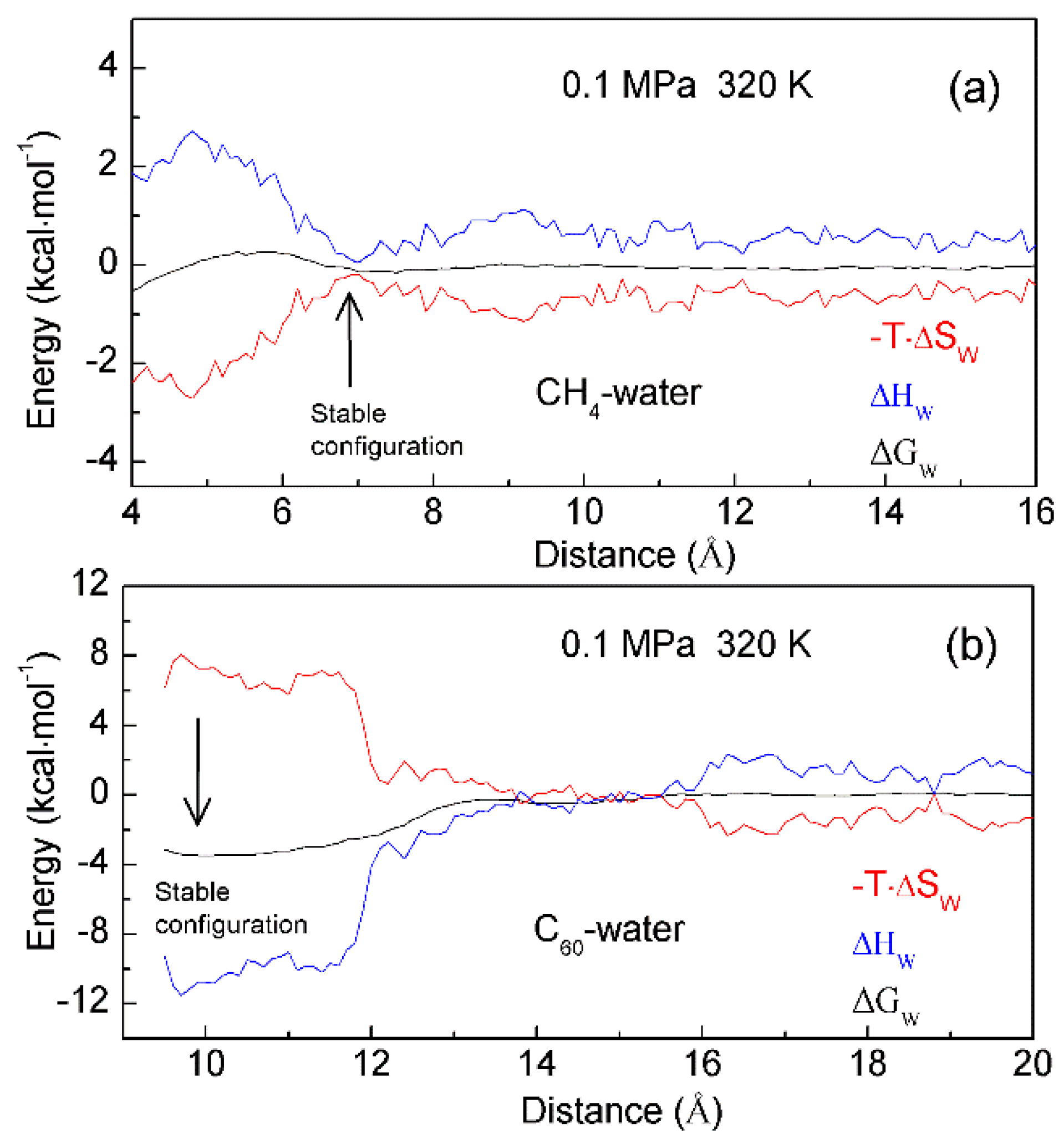
Figure 9. The thermodynamic characteristics of hydrophobic interactions. Based on the calculated PMFs of C60-C60 fullerenes and CH4-CH4 in water at different temperatures (300 K, 320 K and 340 K), water contributions to Gibbs energy (ΔGW) are determined, which are used to calculate the enthalpic (ΔHW) and entropic (-T·ΔSW) contributions. Different thermodynamic characteristics may be expected in initial (a) and hydrophobic (b) solvation processes.
References
- Scheraga, H.A. Theory of hydrophobic interactions. J. Biomol. Struct. Dyn. 1998, 16, 447–460.
- Blokzijl, W.; Engberts, J.B.F.N. Hydrophobic effects. Opinions and facts. Angew. Chem. Int. Ed. 1993, 32, 1545–1579.
- Makowski, M.; Czaplewski, C.; Liwo, A.; Scheraga, H.A. Potential of mean force of association of large hydrophobic particles: Toward the nanoscale limit. J. Phys. Chem. B 2010, 114, 993–1003.
- Sobolewski, E.; Makowski, M.; Czaplewski, C.; Liwo, A.; Ołdziej, S.; Scheraga, H.A. Potential of mean force of hydrophobic association: Dependence on solute size. J. Phys. Chem. B 2007, 111, 10765–10774.
- Bartosik, A.; Wiśniewska, M.; Makowski, M. Potentials of mean force for hydrophobic interactions between hydrocarbons in water solution: Dependence on temperature, solute shape, and solute size. J. Phys. Org. Chem. 2015, 28, 10–16.
- Hansch, C.; Fujita, T. p-σ-π analysis. A method for the correlation of biological activity and chemical structure. J. Am. Chem. Soc. 1964, 86, 1616–1626.
- Kujawski, J.; Popielarska, H.; Myka, A.; Drabińska, B.; Bernard, M.K. The log P parameter as a molecular descriptor in the computer-aided drug design-an overview. Comput. Meth. Sci. Technol. 2012, 18, 81–88.
- Frank, H.S.; Evans, M.W. Free volume and entropy in condensed systems III. Entropy in binary liquid mixtures; partial molal entropy in dilute solutions; structure and thermodynamics in aqueous electrolytes. J. Chem. Phys. 1945, 13, 507–532.
- Davis, J.G.; Gierszal, K.P.; Wang, P.; Ben-Amotz, D. Water structural transformation at molecular hydrophobic interfaces. Nature 2012, 491, 582–585.
- Galamba, N. Water’s structure around hydrophobic solutes and the iceberg model. J. Phys. Chem. B 2013, 117, 2153–2159.
- Raschke, T.M.; Levitt, M. Nonpolar solutes enhance water structure within hydration shells while reducing interactions between them. Proc. Natl. Acad. Sci. USA 2005, 102, 6777–6782.
- Rezus, Y.L.A.; Bakker, H.J. Observation of immobilized water molecules around hydrophobic groups. Phys. Rev. Lett. 2007, 99, 148301.
- Turner, J.; Soper, A.K.; Finney, J.L. A neutron-diffraction study of tetramethylammonium chloride in aqueous solution. Mol. Phys. 1990, 70, 679–700.
- Qvist, J.; Halle, B. Thermal signature of hydrophobic hydration dynamics. J. Am. Chem. Soc. 2008, 130, 10345–10353.
- Buchanan, P.; Aldiwan, N.; Soper, A.K.; Creek, J.L.; Koh, C.A. Decreased structure on dissolving methane in water. Chem. Phys. Lett. 2005, 415, 89–93.
- Bakulin, A.A.; Liang, C.; Jansen, T.L.; Wiersma, D.A.; Bakker, H.J.; Pshenichnikov, M.S. Hydrophobic solvation: A 2D IR spectroscopic inquest. Acc. Chem. Res. 2009, 42, 1229–1238.
- Sun, Q. The effects of dissolved hydrophobic and hydrophilic groups on water structure. J. Solution Chem. 2020, 49, 1473–1484.
- Kauzmann, W. Some factors in the interpretation of protein denaturation. Adv. Protein Chem. 1959, 14, 1–63.
- Ferguson, S.B.; Seward, E.M.; Diederich, F.; Sanford, E.M.; Chou, A.; Inocencio-Szweda, P.; Knobler, C.B. Strong enthalpically driven complexation of neutral benzene guests in aqueous solution. J. Org. Chem. 1988, 53, 5593–5595.
- Baron, R.; Setny, P.; McCammon, J.A. Water in cavity-ligand recognition. J. Am. Chem. Soc. 2010, 132, 12091–12097.
- Setny, P.; Baron, R.; McCammon, J.A. How can hydrophobic association be enthalpy driven? J. Chem. Theory Comput. 2010, 6, 2866–2871.
- Hummer, G. Molecular binding under water’s influence. Nat. Chem. 2010, 2, 906–907.
- Huang, D.M.; Geissler, P.L.; Chandler, D. Scaling of hydrophobic solvation free energies. J. Phys. Chem. B 2001, 105, 6704–6709.
- Lum, K.; Chandler, D.; Weeks, J.D. Hydrophobicity at small and large length scales. J. Phys. Chem. B 1999, 103, 4570–4577.
- Chandler, D. Interfaces and the driving force of hydrophobic assembly. Nature 2005, 437, 640–647.
- Huang, D.M.; Chandler, D. Temperature and length scale dependence of hydrophobic effects and their possible implications for protein folding. Proc. Natl. Acad. Sci. USA 2000, 97, 8324–8327.
- Ball, P. Water as an active constituent in cell biology. Chem. Rev. 2008, 108, 74–108.
- Ball, P. Water is an active matrix of life for cell and molecular biology. Proc. Natl. Acad. Sci. USA 2017, 114, 13327–13335.
- Ball, P. More than a bystander. Nature 2011, 478, 467–468.
- Sun, Q. The Raman OH stretching bands of liquid water. Vib. Spectrosc. 2009, 51, 213–217.
- Sun, Q. Raman spectroscopic study of the effects of dissolved NaCl on water structure. Vib. Spectrosc. 2012, 62, 110–114.
- Sun, Q. Local statistical interpretation for water structure. Chem. Phys. Lett. 2013, 568, 90–94.
- Sun, Q.; Guo, Y. Vibrational sum frequency generation spectroscopy of the air/water interface. J. Mol. Liq. 2016, 213, 28–32.
- Sun, Q. The physical origin of hydrophobic effects. Chem. Phys. Lett. 2017, 672, 21–25.
- Sun, Q.; Zhang, M.X.; Cui, S. The structural origin of hydration repulsive force. Chem. Phys. Lett. 2019, 714, 30–36.
- Sun, Q.; Su, X.W.; Cheng, C.B. The dependence of hydrophobic interactions on the solute size. Chem. Phys. 2019, 516, 199–205.
- Sun, Q.; Wang, W.Q.; Cui, S. Directional nature of hydrophobic interactions: Implications for the mechanism of molecular recognition. Chem. Phys. 2021, 547, 111200.
- Sun, Q.; Cui, S.; Zhang, M.X. Homogeneous nucleation mechanism of NaCl in aqueous solutions. Crystals 2020, 10, 107.
- Stanley, H.E.; Teixeira, J. Interpretation of the unusual behavior of H2O and D2O at low temperatures: Tests of a percolation model. J. Chem. Phys. 1980, 73, 3404–3422.
- Nilsson, A.; Pettersson, L.G.M. Perspective on the structure of liquid water. Chem. Phys. 2011, 389, 1–34.
- Röntgen, W.C. Ueber die Constitution des flüssigen Wassers. Ann. Phys. 1892, 281, 91–97.
- Russo, J.; Tanaka, H. Understanding water’s anomalies with locally favoured structures. Nat. Commun. 2014, 5, 3556.
- Hamm, P. Markov state model of the two-state behaviour of water. J. Chem. Phys. 2016, 145, 134501.
- Shi, R.; Tanaka, H. Microscopic structural descriptor of liquid water. J. Chem. Phys. 2018, 148, 124503.
- Skinner, L.B.; Huang, C.; Schlesinger, D.; Pettersson, L.G.M.; Nilsson, A.; Benmore, C.J. Benchmark oxygen-oxygen pair-distribution function of ambient water from X-ray diffraction measurements with a wide Q-range. J. Chem. Phys. 2013, 138, 074506.
- Hura, G.; Sorenson, J.M.; Glaeser, R.M.; Head-Gordon, T. A high-quality X-ray scattering experiment on liquid water at ambient conditions. J. Chem. Phys. 2000, 113, 9140.
- Misquitta, A.J.; Szalewicz, K. Intermolecular forces from asymptotically corrected density functional description of monomers. Chem. Phys. Lett. 2002, 357, 301–306.
- Misquitta, A.J.; Jeziorski, B.; Szalewicz, K. Dispersion energy from density-functional theory description of monomers. Phys. Rev. Lett. 2003, 91, 033201.
- Hoja, J.; Sax, A.F.; Szalewicz, K. Is electrostatics sufficient to describe hydrogen-bonding interactions? Chem. Eur. J. 2014, 20, 2292–2300.
- Fraley, P.E.; Rao, K.N. High resolution infrared spectra of water vapor: ν1 and ν3 band of H216O. J. Mol. Spectrosc. 1969, 29, 348–364.
- Ludwig, R. The effect of hydrogen bonding on the thermodynamic and spectroscopic properties of molecular clusters and liquids. Phys. Chem. Chem. Phys. 2002, 4, 5481–5487.
- Smith, J.D.; Cappa, C.D.; Wilson, K.R.; Cohen, R.C.; Geissler, P.L.; Saykally, R.J. Unified description of temperature-dependent hydrogen-bond rearrangements in liquid water. Proc. Natl. Acad. Sci. USA 2005, 102, 14171–14174.
- Huang, C.; Wikfeldt, K.T.; Tokushima, T.; Nordlund, D.; Harada, Y.; Bergmann, U.; Niebuhr, M.; Weiss, T.M.; Horikawa, Y.; Leetmaa, M.; et al. The inhomogeneous structure of water at ambient conditions. Proc. Natl. Acad. Sci. USA 2009, 106, 15214–15218.
- Kim, K.H.; Späh, A.; Pathak, H.; Perakis, F.; Mariedahl, D.; Amann-Winkel, K.; Sellberg, J.A.; Lee, J.H.; Kim, S.; Park, J.; et al. Maxima in the thermodynamic response and correlation functions of deeply supercooled water. Science 2017, 358, 1589–1593.
- Matsumoto, M.; Saito, S.; Ohmine, I. Molecular dynamics simulation of the ice nucleation and growth process leading to water freezing. Nature 2002, 416, 409–413.
- Moore, E.B.; Molinero, V. Structural transformation in supercooled water controls the crystallization rate of ice. Nature 2011, 479, 506–508.
- Fitzner, M.; Sosso, G.C.; Cox, S.J.; Michaelides, A. Ice is born in low-mobility regions of supercooled liquid water. Proc. Natl. Acad. Sci. USA 2019, 116, 2009–2014.
- Trudu, F.; Donadio, D.; Parrinello, M. Freezing of a Lennard-Jones fluid: From nucleation to spinodal regime. Phys. Rev. Lett. 2006, 97, 105701.
- Berryman, J.T.; Anwar, M.; Dorosz, S.; Schilling, T. The early crystal nucleation process in hard spheres shows synchronised ordering and densification. J. Chem. Phys. 2016, 145, 211901.
- Desgranges, C.; Delhommelle, J. Can ordered precursors promote the nucleation of solid solutions? Phys. Rev. Lett. 2019, 123, 195701.
- Speedy, R.J. Limiting forms of the thermodynamic divergences at the conjectured stability limits in superheated and supercooled water. J. Phys. Chem. 1982, 86, 3002–3005.
- Poole, P.H.; Sciortino, F.; Essmann, U.; Stanley, H.E. Phase behaviour of metastable water. Nature 1992, 360, 324–328.
- Sastry, S.; Debenedetti, P.G.; Sciortino, F.; Stanley, H.E. Singularity-free interpretation of the thermodynamics of supercooled water. Phys. Rev. E 1996, 53, 6144.
- Angell, C.A. Insights into phases of liquid water from study of its unusual glass-forming properties. Science 2008, 319, 582–587.
- Kim, K.H.; Amann-Winkel, K.; Giovambattista, N.; Späh, A.; Perakis, F.; Pathak, H.; Parada, M.L.; Yang, C.; Mariedahl, D.; Eklund, T.; et al. Experimental observation of the liquid-liquid transition in bulk supercooled water under pressure. Science 2020, 370, 978–982.
- Palmer, J.C.; Martelli, F.; Liu, Y.; Car, R.; Panagiotopoulos, A.Z.; Debenedetti, P.G. Metastable liquid-liquid transition in a molecular model of water. Nature 2014, 510, 385–388.
- Debenedetti, P.G.; Sciortino, F.; Zerze, G.H. Second critical point in two realistic models of water. Science 2020, 369, 289–292.
- Gartner, T.E.; Zhang, L.; Piaggi, P.M.; Car, R.; Panagiotopoulos, A.Z.; Debenedetti, P.G. Signatures of a liquid-liquid transition in an ab initio deep neural network model for water. Proc. Natl. Acad. Sci. USA 2020, 117, 26040–26046.
- Handle, P.H.; Loerting, T.; Sciortino, F. Supercooled and glassy water: Metastable liquid(s), amorphous solid(s), and a no-man’s land. Proc. Natl. Acad. Sci. USA 2017, 114, 13336–13344.
- Collins, K.D.; Neilson, G.W.; Enderby, J.E. Ions in water: Characterizing the forces that control chemical processes and biological structure. Biophys. Chem. 2007, 128, 95–104.
- Cappa, C.D.; Smith, J.D.; Messer, B.M.; Cohen, R.C.; Saykally, R.J. Effects of cations on the hydrogen bond network of liquid water: New results from X-ray absorption spectroscopy of liquid microjets. J. Phys. Chem. B 2006, 110, 5301–5309.
- Omta, A.W.; Kropman, M.F.; Woutersen, S.; Bakker, H.J. Negligible effect of ions on the hydrogen-bond structure in liquid water. Science 2003, 301, 347–349.
- Moilanen, D.E.; Wong, D.; Rosenfeld, D.E.; Fenn, E.E.; Fayer, M.D. Ion-water hydrogen-bond switching observed with 2D IR vibrational echo chemical exchange spectroscopy. Proc. Natl. Acad. Sci. USA 2009, 106, 375–380.
- Turton, D.A.; Hunger, J.; Hefter, G.; Buchner, R.; Wynne, K. Glasslike behavior in aqueous electrolyte solutions. J. Chem. Phys. 2008, 128, 161102.
- Thompson, H.; Soper, A.K.; Ricci, M.A.; Bruni, F.; Skipper, N.T. The three-dimensional structure of water confined in nanoporous vycor glass. J. Phys. Chem. B 2007, 111, 5610–5620.
- Giri, A.K.; Teixeira, F.; Cordeiro, M.N.D.S. Structure and kinetics of water in highly confined conditions: A molecular dynamics simulation study. J. Mol. Liq. 2018, 268, 625–636.
- Winarto; Takaiwa, D.; Yamamoto, E.; Yasuoka, K. Structures of water molecules in carbon nanotubes under electric fields. J. Chem. Phys. 2015, 142, 124701.
- Dorsey, N.E. Properties of Ordinary Water Substance; ACS Monograph No. 81; Reinhold Publishing Corp.: New York, NY, USA, 1940.
- Chodera, J.D.; Mobley, D.L. Entropy-enthalpy compensation: Role and ramifications in biomolecular ligand recognition and design. Annu. Rev. Biophys. 2013, 42, 121–142.
- Dragan, A.I.; Read, C.M.; Crane-Robinson, C. Enthalpy-entropy compensation: The role of solvation. Eur. Biophys. J. 2017, 46, 301–308.
- Breiten, B.; Lockett, M.R.; Sherman, W.; Fujita, S.; Al-Sayah, M.; Lange, H.; Bowers, C.M.; Heroux, A.; Krilov, G.; Whitesides, G.M. Water networks contribute to enthalpy/entropy compensation in protein-ligand binding. J. Am. Chem. Soc. 2013, 135, 15579–15584.
- Sharp, K. Entropy-enthalpy compensation: Fact or artifact? Protein Sci. 2001, 10, 661–667.
- Gilli, P.; Ferrett, V.; Gilli, G.; Borea, P.A. Enthalpy-entropy compensation in drug-receptor binding. J. Phys. Chem. 1994, 98, 1515–1518.
More