 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Vivi Li | -- | 1346 | 2022-10-26 01:48:57 |
Video Upload Options
A nucleophilic substitution is a class of chemical reactions in which an electron-rich chemical species (known as a nucleophile) replaces a functional group within another electron-deficient molecule (known as the electrophile). The molecule that contains the electrophile and the leaving functional group is called the substrate. The most general form of the reaction may be given as the following: The electron pair (:) from the nucleophile (Nuc) attacks the substrate (R-LG) and bonds with it. Simultaneously, the leaving group (LG) departs with an electron pair. The principal product in this case is R-Nuc. The nucleophile may be electrically neutral or negatively charged, whereas the substrate is typically neutral or positively charged. An example of nucleophilic substitution is the hydrolysis of an alkyl bromide, R-Br under basic conditions, where the attacking nucleophile is OH− and the leaving group is Br−. Nucleophilic substitution reactions are common in organic chemistry (especially introductory organic chemistry). Nucleophiles often attack a saturated aliphatic carbon. Less often, they may attack an aromatic or unsaturated carbon.
1. Saturated Carbon Centres
1.1. SN1 and SN2 Reactions
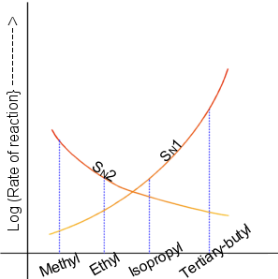
In 1935, Edward D. Hughes and Sir Christopher Ingold studied nucleophilic substitution reactions of alkyl halides and related compounds. They proposed that there were two main mechanisms at work, both of them competing with each other. The two main mechanisms were the SN1 reaction and the SN2 reaction, where S stands for substitution, N stands for nucleophilic, and the number represents the kinetic order of the reaction.[1]
In the SN2 reaction, the addition of the nucleophile and the elimination of leaving group take place simultaneously (i.e. a concerted reaction). SN2 occurs when the central carbon atom is easily accessible to the nucleophile.[2]
| Nucleophilic substitution at carbon | |
|---|---|
|
mechanism. https://handwiki.org/wiki/index.php?curid=1181387 |
|
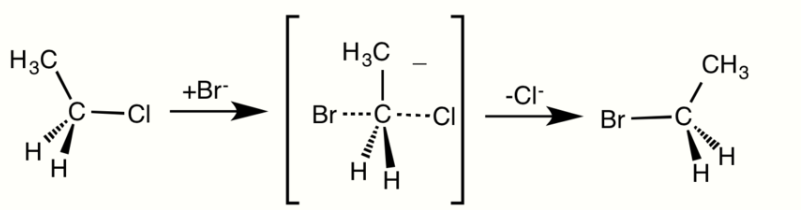
In SN2 reactions, there are a few conditions that affect the rate of the reaction. First of all, the 2 in SN2 implies that there are two concentrations of substances that affect the rate of reaction: substrate (Sub) and nucleophile. The rate equation for this reaction would be Rate=k[Sub][Nuc]. For a SN2 reaction, an aprotic solvent is best, such as acetone, DMF, or DMSO. Aprotic solvents do not add protons (H+ ions) into solution; if protons were present in SN2 reactions, they would react with the nucleophile and severely limit the reaction rate. Since this reaction occurs in one step, steric effects drive the reaction speed. In the intermediate step, the nucleophile is 185 degrees from the leaving group and the stereochemistry is inverted as the nucleophile bonds to make the product. Also, because the intermediate is partially bonded to the nucleophile and leaving group, there is no time for the substrate to rearrange itself: the nucleophile will bond to the same carbon that the leaving group was attached to. A final factor that affects reaction rate is nucleophilicity; the nucleophile must attack an atom other than a hydrogen.
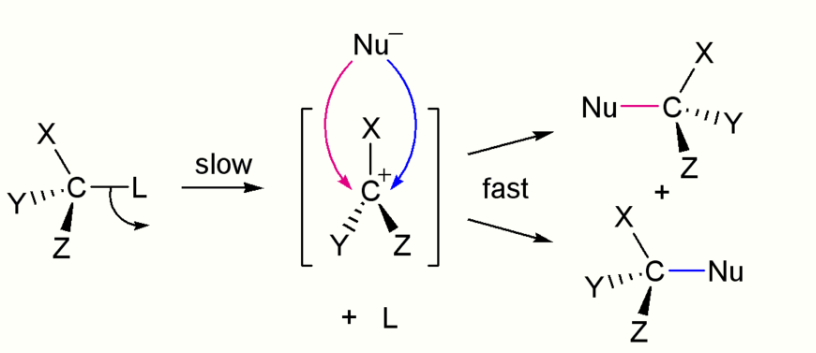
By contrast the SN1 reaction involves two steps. SN1 reactions tend to be important when the central carbon atom of the substrate is surrounded by bulky groups, both because such groups interfere sterically with the SN2 reaction (discussed above) and because a highly substituted carbon forms a stable carbocation.
| Nucleophilic substitution at carbon | |
|---|---|
 |
|
| SN1 mechanism. https://handwiki.org/wiki/index.php?curid=1448860 | |
Like SN2 reactions, there are quite a few factors that affect the reaction rate of SN1 reactions. Instead of having two concentrations that affect the reaction rate, there is only one, substrate. The rate equation for this would be Rate=k[Sub]. Since the rate of a reaction is only determined by its slowest step, the rate at which the leaving group "leaves" determines the speed of the reaction. This means that the better the leaving group, the faster the reaction rate. A general rule for what makes a good leaving group is the weaker the conjugate base, the better the leaving group. In this case, halogens are going to be the best leaving groups, while compounds such as amines, hydrogen, and alkanes are going to be quite poor leaving groups. As SN2 reactions were affected by sterics, SN1 reactions are determined by bulky groups attached to the carbocation. Since there is an intermediate that actually contains a positive charge, bulky groups attached are going to help stabilize the charge on the carbocation through resonance and distribution of charge. In this case, tertiary carbocation will react faster than a secondary which will react much faster than a primary. It is also due to this carbocation intermediate that the product does not have to have inversion. The nucleophile can attack from the top or the bottom and therefore create a racemic product. It is important to use a protic solvent, water and alcohols, since an aprotic solvent could attack the intermediate and cause unwanted product. It does not matter if the hydrogens from the protic solvent react with the nucleophile since the nucleophile is not involved in the rate determining step.
| Table 1. Nucleophilic substitutions on RX (an alkyl halide or equivalent) | |||||||
|---|---|---|---|---|---|---|---|
| Factor | SN1 | SN2 | Comments | ||||
| Kinetics | Rate = k[RX] | Rate = k[RX][Nuc] | |||||
| Primary alkyl | Never unless additional stabilising groups present | Good unless a hindered nucleophile is used | |||||
| Secondary alkyl | Moderate | Moderate | |||||
| Tertiary alkyl | Excellent | Never | Elimination likely if heated or if strong base used | ||||
| Leaving group | Important | Important | For halogens, I > Br > Cl >> F |
||||
| Nucleophilicity | Unimportant | Important | |||||
| Preferred solvent | Polar protic | Polar aprotic | |||||
| Stereochemistry | Racemisation (+ partial inversion possible) | Inversion | |||||
| Rearrangements | Common | Rare | Side reaction | ||||
| Eliminations | Common, especially with basic nucleophiles | Only with heat & basic nucleophiles | Side reaction esp. if heated |
||||
2. Reactions
There are many reactions in organic chemistry involve this type of mechanism. Common examples include:
- Organic reductions with hydrides, for example
-
- R-X → R-H using LiAlH4 (SN2
- Hydrolysis reactions such as
-
- R-Br + OH− → R-OH + Br− (SN2) or
- R-Br + H2O → R-OH + HBr (SN1)
- Williamson ether synthesis
-
- R-Br + OR'− → R-OR' + Br− (SN2)
- The Wenker synthesis, a ring-closing reaction of aminoalcohols.
- The Finkelstein reaction, a halide exchange reaction. Phosphorus nucleophiles appear in the Perkow reaction and the Michaelis–Arbuzov reaction.
- The Kolbe nitrile synthesis, the reaction of alkyl halides with cyanides.
2.1. Borderline Mechanism
An example of a substitution reaction taking place by a so-called borderline mechanism as originally studied by Hughes and Ingold[3] is the reaction of 1-phenylethyl chloride with sodium methoxide in methanol.
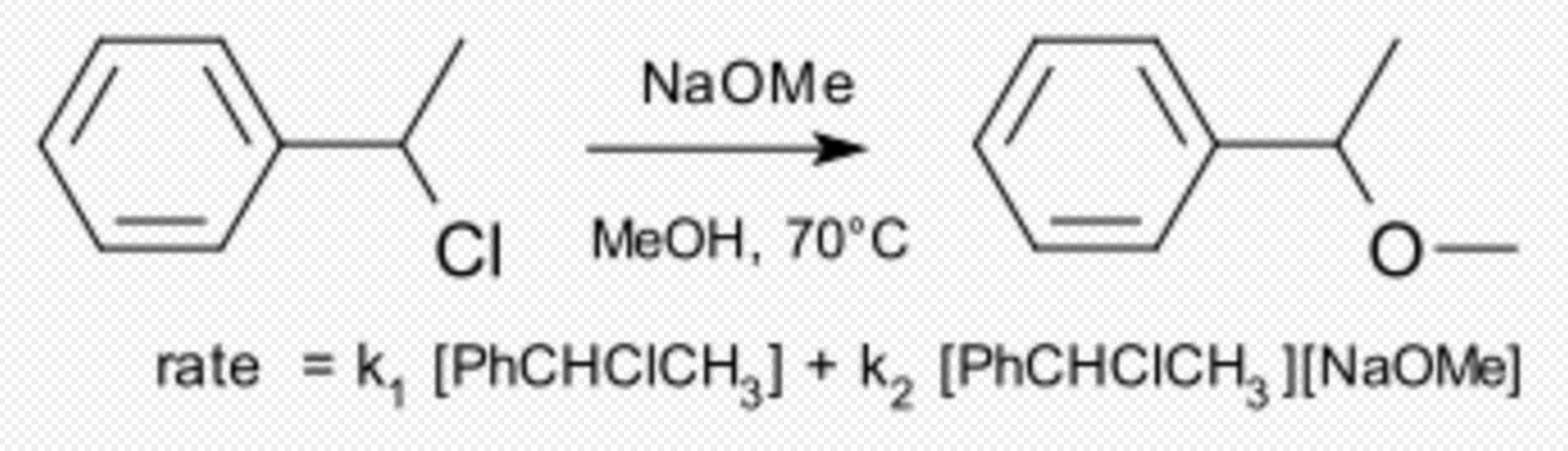
The reaction rate is found to the sum of SN1 and SN2 components with 61% (3,5 M, 70 °C) taking place by the latter. https://handwiki.org/wiki/index.php?curid=2040441
2.2. Other Mechanisms
Besides SN1 and SN2, other mechanisms are known, although they are less common. The SNi mechanism is observed in reactions of thionyl chloride with alcohols, and it is similar to SN1 except that the nucleophile is delivered from the same side as the leaving group.
Nucleophilic substitutions can be accompanied by an allylic rearrangement as seen in reactions such as the Ferrier rearrangement. This type of mechanism is called an SN1' or SN2' reaction (depending on the kinetics). With allylic halides or sulphonates, for example, the nucleophile may attack at the γ unsaturated carbon in place of the carbon bearing the leaving group. This may be seen in the reaction of 1-chloro-2-butene with sodium hydroxide to give a mixture of 2-buten-1-ol and 1-buten-3-ol:
- CH3CH=CH-CH2-Cl → CH3CH=CH-CH2-OH + CH3CH(OH)-CH=CH2
The Sn1CB mechanism appears in inorganic chemistry. Competing mechanisms exist.[4][5]
In organometallic chemistry the nucleophilic abstraction reaction occurs with a nucleophilic substitution mechanism.
3. Unsaturated Carbon Centres
Nucleophilic substitution via the SN1 or SN2 mechanism does not generally occur with vinyl or aryl halides or related compounds. Under certain conditions nucleophilic substitutions may occur, via other mechanisms such as those described in the nucleophilic aromatic substitution article.
When the substitution occurs at the carbonyl group, the acyl group may undergo nucleophilic acyl substitution. This is the normal mode of substitution with carboxylic acid derivatives such as acyl chlorides, esters and amides.
References
- S. R. Hartshorn, Aliphatic Nucleophilic Substitution, Cambridge University Press, London, 1973. ISBN:0-521-09801-7
- Introducing Aliphatic Substitution with a Discovery Experiment Using Competing Electrophiles Timothy P. Curran, Amelia J. Mostovoy, Margaret E. Curran, and Clara Berger Journal of Chemical Education 2016 93 (4), 757-761 doi:10.1021/acs.jchemed.5b00394 https://doi.org/10.1021%2Facs.jchemed.5b00394
- 253. Reaction kinetics and the Walden inversion. Part II. Homogeneous hydrolysis, alcoholysis, and ammonolysis of -phenylethyl halides Edward D. Hughes, Christopher K. Ingold and Alan D. Scott, J. Chem. Soc., 1937, 1201 doi:10.1039/JR9370001201 https://doi.org/10.1039%2FJR9370001201
- N.S.Imyanitov. Electrophilic Bimolecular Substitution as an Alternative to Nucleophilic Monomolecular Substitution in Inorganic and Organic Chemistry. J. Gen. Chem. USSR (Engl. Transl.) 1990; 60 (3); 417-419.
- Unimolecular Nucleophilic Substitution does not Exist! / N.S.Imyanitov. SciTecLibrary http://sciteclibrary.ru/eng/catalog/pages/9330.html

