 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Qianyu He | -- | 1992 | 2022-10-26 04:16:59 | | | |
| 2 | Qianyu He | + 126 word(s) | 2118 | 2022-10-26 04:21:42 | | | | |
| 3 | Catherine Yang | Meta information modification | 2118 | 2022-10-26 05:41:42 | | | | |
| 4 | Catherine Yang | Meta information modification | 2118 | 2022-10-26 05:42:34 | | |
Video Upload Options
Pancreatic cancer is one of the most intractable malignant tumors worldwide, and is known for its refractory and poor prognosis. Pancreatic ductal adenocarcinoma (PDAC) is the most common type of pancreatic cancer. KRAS is the most commonly mutated oncogene in PDAC. It has been considered the “untargetable” oncogene for decades until the emergence of G12C inhibitors, which put an end to this dilemma by covalent binding to the switch-II pocket of the G12C mutant protein. However, G12C inhibitors showed remarkable efficacy against non-small-cell lung cancer (NSCLC), while the G12C mutation is rare in PDAC. Based on the successful experience of G12C inhibitors, targeting KRAS G12D/V, which forms the majority of KRAS mutations in PDAC, is gradually being regarded as a potential therapy.
1. KRAS Mutations in PDAC
1.1. Molecular Mechanism of KRAS Mutations
1.2. Progress of PDAC with KRAS Mutations
2. KRAS Inhibitors for PDAC
2.1. KRASG12C Inhibitors
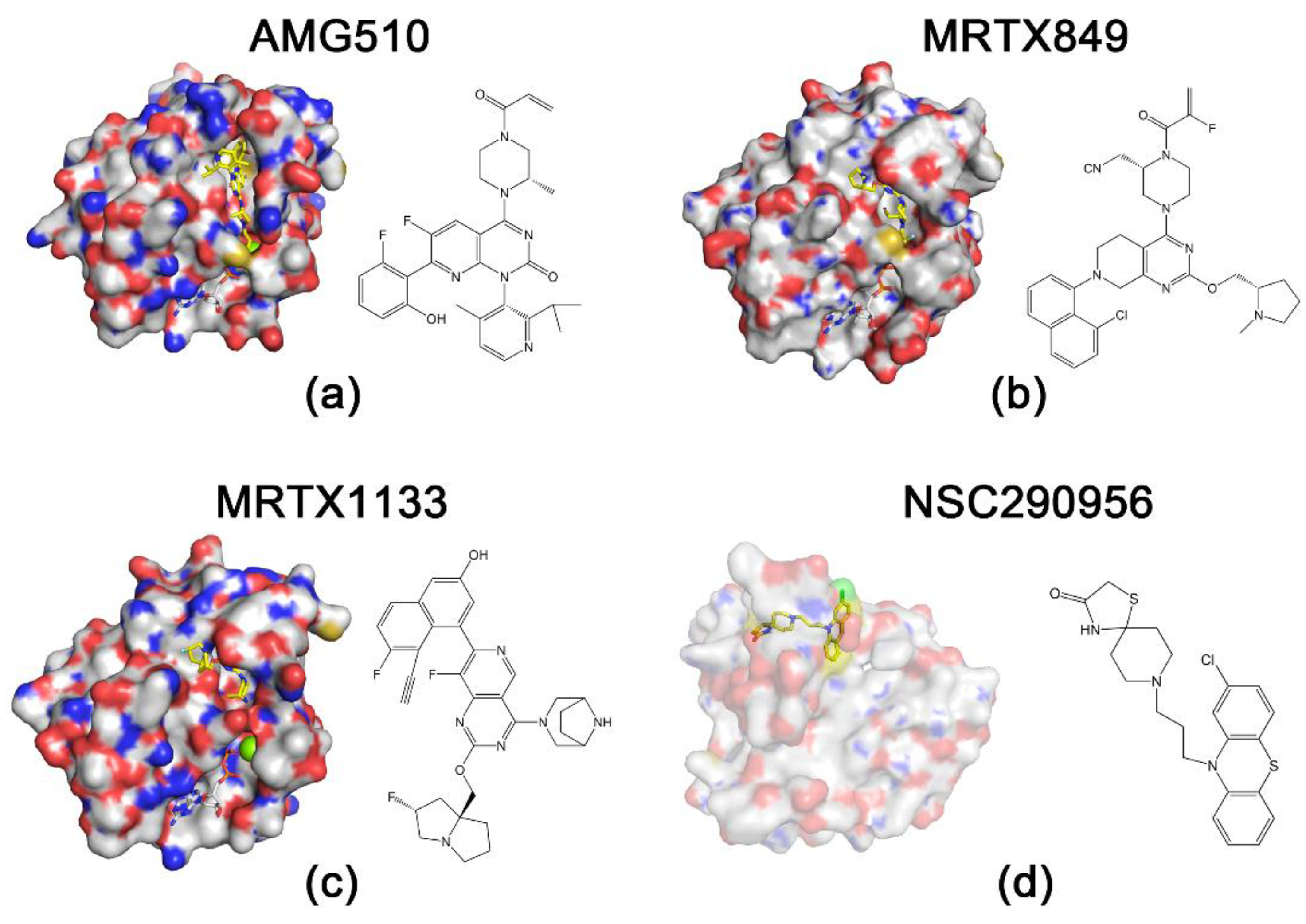
2.2. KRASG12D Inhibitors
2.2.1. MRTX1133
2.2.2. Peptide Nucleic Acids (PNAs)
2.3. Pan-RAS Inhibitors
References
- Soh, J.; Okumura, N.; Lockwood, W.W.; Yamamoto, H.; Shigematsu, H.; Zhang, W.; Chari, R.; Shames, D.S.; Tang, X.; MacAulay, C.; et al. Oncogene mutations, copy number gains and mutant allele specific imbalance (MASI) frequently occur together in tumor cells. PLoS ONE 2009, 4, e7464.
- Moore, A.R.; Rosenberg, S.C.; McCormick, F.; Malek, S. Author Correction: RAS-targeted therapies: Is the undruggable drugged? Nat. Rev. Drug Discov. 2020, 19, 902.
- The Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337.
- The Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550.
- The Cancer Genome Atlas Research Network. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell. 2017, 32, 185–203.e13.
- Takai, Y.; Sasaki, T.; Matozaki, T. Small GTP-binding proteins. Physiol. Rev. 2001, 81, 153–208.
- Drugan, J.K.; Rogers-Graham, K.; Gilmer, T.; Campbell, S.; Clark, G.J. The Ras/p120 GTPase-activating protein (GAP) interaction is regulated by the p120 GAP pleckstrin homology domain. J. Biol. Chem. 2000, 275, 35021–35027.
- Bos, J.L.; Rehmann, H.; Wittinghofer, A. GEFs and GAPs: Critical elements in the control of small G proteins. Cell 2007, 129, 865–877.
- Pamonsinlapatham, P.; Hadj-Slimane, R.; Lepelletier, Y.; Allain, B.; Toccafondi, M.; Garbay, C.; Raynaud, F. p120-Ras GTPase activating protein (RasGAP): A multi-interacting protein in downstream signaling. Biochimie 2009, 91, 320–328.
- Hunter, J.C.; Manandhar, A.; Carrasco, M.A.; Gurbani, D.; Gondi, S.; Westover, K.D. Biochemical and Structural Analysis of Common Cancer-Associated KRAS Mutations. Mol. Cancer Res. 2015, 13, 1325–1335.
- Ostrem, J.M.; Shokat, K.M. Direct small-molecule inhibitors of KRAS: From structural insights to mechanism-based design. Nat. Rev. Drug Discov. 2016, 15, 771–785.
- Rabara, D.; Tran, T.H.; Dharmaiah, S.; Stephens, R.M.; McCormick, F.; Simanshu, D.K.; Holderfield, M. KRAS G13D sensitivity to neurofibromin-mediated GTP hydrolysis. Proc. Natl. Acad. Sci. USA 2019, 116, 22122–22131.
- Huang, L.; Jansen, L.; Balavarca, Y.; Babaei, M.; van der Geest, L.; Lemmens, V.; Van Eycken, L.; De Schutter, H.; Johannesen, T.B.; Primic-Zakelj, M.; et al. Stratified survival of resected and overall pancreatic cancer patients in Europe and the USA in the early twenty-first century: A large, international population-based study. BMC Med. 2018, 16, 125.
- Ostrem, J.M.; Peters, U.; Sos, M.L.; Wells, J.A.; Shokat, K.M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548–551.
- Ambrogio, C.; Kohler, J.; Zhou, Z.W.; Wang, H.; Paranal, R.; Li, J.; Capelletti, M.; Caffarra, C.; Li, S.; Lv, Q.; et al. KRAS Dimerization Impacts MEK Inhibitor Sensitivity and Oncogenic Activity of Mutant KRAS. Cell 2018, 172, 857–868.e15.
- Itonaga, M.; Ashida, R.; Murata, S.I.; Yamashita, Y.; Hatamaru, K.; Tamura, T.; Kawaji, Y.; Kayama, Y.; Emori, T.; Kawai, M.; et al. Kras Gene Analysis Using Liquid-Based Cytology Specimens Predicts Therapeutic Responses and Prognosis in Patients with Pancreatic Cancer. Cancers 2022, 14, 551.
- McIntyre, C.A.; Lawrence, S.A.; Richards, A.L.; Chou, J.F.; Wong, W.; Capanu, M.; Berger, M.F.; Donoghue, M.T.A.; Yu, K.H.; Varghese, A.M.; et al. Alterations in driver genes are predictive of survival in patients with resected pancreatic ductal adenocarcinoma. Cancer 2020, 126, 3939–3949.
- Dong, L.; Wang, S.; Fu, B.; Wang, J. Evaluation of droplet digital PCR and next generation sequencing for characterizing DNA reference material for KRAS mutation detection. Sci. Rep. 2018, 8, 9650.
- Lin, M.T.; Mosier, S.L.; Thiess, M.; Beierl, K.F.; Debeljak, M.; Tseng, L.H.; Chen, G.; Yegnasubramanian, S.; Ho, H.; Cope, L.; et al. Clinical validation of KRAS, BRAF, and EGFR mutation detection using next-generation sequencing. Am. J. Clin. Pathol. 2014, 141, 856–866.
- Haque, M.R.; Wessel, C.R.; Leary, D.D.; Wang, C.; Bhushan, A.; Bishehsari, F. Patient-derived pancreatic cancer-on-a-chip recapitulates the tumor microenvironment. Microsyst. Nanoeng. 2022, 8, 36.
- Nauheim, D.; Moskal, D.; Renslo, B.; Chadwick, M.; Jiang, W.; Yeo, C.J.; Nevler, A.; Bowne, W.; Lavu, H. KRAS mutation allele frequency threshold alters prognosis in right-sided resected pancreatic cancer. J. Surg. Oncol. 2022, 126, 314–321.
- Dopico, P.J.; Le, M.N.; Burgess, B.; Yang, Z.; Zhao, Y.; Wang, Y.; George, T.J.; Fan, Z.H. Longitudinal Study of Circulating Biomarkers in Patients with Resectable Pancreatic Ductal Adenocarcinoma. Biosensors 2022, 12, 206.
- Li, Z.; Zhuang, H.; Chen, X.; Zhang, Y.; Ma, Z.; Wang, S.; Yan, Q.; Zhou, Z.; Huang, S.; Zhang, C.; et al. Identification of MBOAT2 as an Unfavorable Biomarker Correlated with KRAS Activation and Reduced CD8(+) T-Cell Infiltration in Pancreatic Cancer. J. Oncol. 2022, 2022, 4269733.
- Fakih, M.; O’Neil, B.; Price, T.J.; Falchook, G.S.; Desai, J.; Kuo, J.; Govindan, R.; Rasmussen, E.; Morrow, P.K.H.; Ngang, J.; et al. Phase 1 study evaluating the safety, tolerability, pharmacokinetics (PK), and efficacy of AMG 510, a novel small molecule KRAS(G12c) inhibitor, in advanced solid tumors. Meeting Abstract. J. Clin. Oncol. 2019, 37, 3003.
- Hallin, J.; Engstrom, L.D.; Hargis, L.; Calinisan, A.; Aranda, R.; Briere, D.M.; Sudhakar, N.; Bowcut, V.; Baer, B.R.; Ballard, J.A.; et al. The KRAS(G12C) Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov. 2020, 10, 54–71.
- Brauswetter, D.; Gurbi, B.; Varga, A.; Varkondi, E.; Schwab, R.; Banhegyi, G.; Fabian, O.; Keri, G.; Valyi-Nagy, I.; Petak, I. Molecular subtype specific efficacy of MEK inhibitors in pancreatic cancers. PLoS ONE 2017, 12, e0185687.
- Zhou, L.; Baba, Y.; Kitano, Y.; Miyake, K.; Zhang, X.; Yamamura, K.; Kosumi, K.; Kaida, T.; Arima, K.; Taki, K.; et al. KRAS, BRAF, and PIK3CA mutations, and patient prognosis in 126 pancreatic cancers: Pyrosequencing technology and literature review. Med. Oncol. 2016, 33, 32.
- Zhang, J.; Liu, Z.; Zhao, W.; Yin, X.; Zheng, X.; Liu, C.; Wang, J.; Wang, E. Discovery of Small Molecule NSC290956 as a Therapeutic Agent for KRas Mutant Non-Small-Cell Lung Cancer. Front. Pharmacol. 2021, 12, 797821.
- Nakajima, E.C.; Drezner, N.; Li, X.; Mishra-Kalyani, P.S.; Liu, Y.; Zhao, H.; Bi, Y.; Liu, J.; Rahman, A.; Wearne, E.; et al. FDA Approval Summary: Sotorasib for KRAS G12C-Mutated Metastatic NSCLC. Clin. Cancer Res. 2022, 28, 1482–1486.
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223.
- Fell, J.B.; Fischer, J.P.; Baer, B.R.; Blake, J.F.; Bouhana, K.; Briere, D.M.; Brown, K.D.; Burgess, L.E.; Burns, A.C.; Burkard, M.R.; et al. Identification of the Clinical Development Candidate MRTX849, a Covalent KRAS(G12C) Inhibitor for the Treatment of Cancer. J. Med. Chem. 2020, 63, 6679–6693.
- Hansen, R.; Peters, U.; Babbar, A.; Chen, Y.; Feng, J.; Janes, M.R.; Li, L.S.; Ren, P.; Liu, Y.; Zarrinkar, P.P. The reactivity-driven biochemical mechanism of covalent KRAS(G12C) inhibitors. Nat. Struct. Mol. Biol. 2018, 25, 454–462.
- Vasta, J.D.; Peacock, D.M.; Zheng, Q.; Walker, J.A.; Zhang, Z.; Zimprich, C.A.; Thomas, M.R.; Beck, M.T.; Binkowski, B.F.; Corona, C.R.; et al. KRAS is vulnerable to reversible switch-II pocket engagement in cells. Nat. Chem. Biol. 2022, 18, 596–604.
- Wang, X.; Allen, S.; Blake, J.F.; Bowcut, V.; Briere, D.M.; Calinisan, A.; Dahlke, J.R.; Fell, J.B.; Fischer, J.P.; Gunn, R.J.; et al. Identification of MRTX1133, a Noncovalent, Potent, and Selective KRAS(G12D) Inhibitor. J. Med. Chem. 2022, 65, 3123–3133.
- Pellestor, F.; Paulasova, P. The peptide nucleic acids (PNAs), powerful tools for molecular genetics and cytogenetics. Eur. J. Hum. Genet. 2004, 12, 694–700.
- Dean, D.A. Peptide nucleic acids: Versatile tools for gene therapy strategies. Adv. Drug Deliv. Rev. 2000, 44, 81–95.
- De Cola, C.; Manicardi, A.; Corradini, R.; Izzo, I.; De Riccardis, F. Carboxyalkyl peptoid PNAs: Synthesis and hybridization properties. Article. Tetrahedron 2012, 68, 499–506.
- Chiarantini, L.; Cerasi, A.; Fraternale, A.; Millo, E.; Benatti, U.; Sparnacci, K.; Laus, M.; Ballestri, M.; Tondelli, L. Comparison of novel delivery systems for antisense peptide nucleic acids. J. Control. Release 2005, 109, 24–36.
- Shai, A.; Galouk, E.; Miari, R.; Tareef, H.; Sammar, M.; Zeidan, M.; Rayan, A.; Falah, M. Inhibiting mutant KRAS G12D gene expression using novel peptide nucleic acid-based antisense: A potential new drug candidate for pancreatic cancer. Oncol. Lett. 2022, 23, 130.
- Coley, A.B.; Ward, A.; Keeton, A.B.; Chen, X.; Maxuitenko, Y.; Prakash, A.; Li, F.; Foote, J.B.; Buchsbaum, D.J.; Piazza, G.A. Pan-RAS inhibitors: Hitting multiple RAS isozymes with one stone. Adv. Cancer Res. 2022, 153, 131–168.
- Keeton, A.B.; Ward, A.; Chen, X.; Valiyaveettil, J.; Zhu, B.; Ramirez-Alcantara, V. Abstract 2707: A novel RAS inhibitor, MCI-062, inhibits colon tumor growth in vivo and activates antitumor immunity. Cancer Res. 2019, 79, 2707.
- Welsch, M.E.; Kaplan, A.; Chambers, J.M.; Stokes, M.E.; Bos, P.H.; Zask, A.; Zhang, Y.; Sanchez-Martin, M.; Badgley, M.A.; Huang, C.S.; et al. Multivalent Small-Molecule Pan-RAS Inhibitors. Cell 2017, 168, 878–889.e29.
- Rognan, D. Rational design of protein–protein interaction inhibitors. Med. Chem. Commun. 2015, 6, 51–60.
- Khan, I.; Rhett, J.M.; O’Bryan, J.P. Therapeutic targeting of RAS: New hope for drugging the “undruggable”. Biochim. Biophys. Acta. Mol. Cell Res. 2020, 1867, 118570.
- Gorgulla, C.; Boeszoermenyi, A.; Wang, Z.F.; Fischer, P.D.; Coote, P.W.; Padmanabha Das, K.M.; Malets, Y.S.; Radchenko, D.S.; Moroz, Y.S.; Scott, D.A.; et al. An open-source drug discovery platform enables ultra-large virtual screens. Nature 2020, 580, 663–668.
- Zheng, X.; Liu, Z.; Li, D.; Wang, E.; Wang, J. Rational drug design: The search for Ras protein hydrolysis intermediate conformation inhibitors with both affinity and specificity. Curr. Pharm. Des. 2013, 19, 2246–2258.
- Guo, N.; Liu, Z.; Zhao, W.; Wang, E.; Wang, J. Small Molecule APY606 Displays Extensive Antitumor Activity in Pancreatic Cancer via Impairing Ras-MAPK Signaling. PLoS ONE 2016, 11, e0155874.
- Guo, X.; Zhao, W.; Liu, Z.; Wang, J. Spiclomazine displays a preferential anti-tumor activity in mutant KRas-driven pancreatic cancer. Oncotarget 2018, 9, 6938–6951.
- Zhao, W.; Li, D.; Liu, Z.; Zheng, X.; Wang, J.; Wang, E. Spiclomazine induces apoptosis associated with the suppression of cell viability, migration and invasion in pancreatic carcinoma cells. PLoS ONE 2013, 8, e66362.
- Li, D.; Liu, Z.; Zhao, W.; Zheng, X.; Wang, J.; Wang, E. A small-molecule induces apoptosis and suppresses metastasis in pancreatic cancer cells. Eur. J. Pharm. Sci. 2013, 48, 658–667.
- Liu, Z.; Li, D.; Zheng, X.; Wang, E.; Wang, J. Selective induction of apoptosis: Promising therapy in pancreatic cancer. Curr. Pharm. Des. 2013, 19, 2259–2268.
- Liu, Z.; Li, D.; Zhao, W.; Zheng, X.; Wang, J.; Wang, E. A potent lead induces apoptosis in pancreatic cancer cells. PLoS ONE 2012, 7, e37841.

