1. KRAS Mutations in PDAC
RAS (rat sarcoma virus) genes constitute one of the most commonly mutated gene families in malignant tumors [
52]. The RAS gene family includes three genes: KRAS, HRAS and NRAS. KRAS is the most common mutation type of the RAS gene, accounting for 80% of RAS gene-related malignancies. The KRAS gene encodes two splice variants using different exon 4 s, producing KRAS4A and KRAS4B. It has been experimentally demonstrated that both the isoforms are associated with tumor formation [
11]. KRAS mutations have mainly been found in lung cancer (32%), PDAC (86%), and colon cancer (41%) [
53,
54,
55]. The most common isoforms of KRAS in PDAC are KRAS
G12D (45%) and KRAS
G12V (35%) [
56].
1.1. Molecular Mechanism of KRAS Mutations
From the perspective of function, the protein expressed by the KRAS gene is a purine nucleotide binding protein located on the cell membrane and has the activity of GTPase [
57]. KRAS protein, as a binary switch of guanosine diphosphate (GDP)/guanosine triphosphate (GTP), controls important signal transduction from activated membrane receptors to intracellular molecules [
58]. In the inactive state, KRAS protein binds to GDP [
59]. When stimulated by relevant signal molecules (such as epidermal growth factor receptor EGFR), the binding ability of KRAS protein to GDP is weakened. GTP takes the place of GDP to bind to the RAS protein, and the KRAS protein is, therefore, activated to bind with downstream signal molecules as monomers or dimers for signal transduction. Then, with the effect of GTP-activated proteins (GAPs), the GTPase activity of KRAS is significantly increased, and GTP combined with KRAS is hydrolyzed into GDP, restoring KRAS to its inactivated state [
60]. However, in tumor cells, KRAS gene mutation leads to the loss of GTPase activity in the KRAS protein, which makes it unable to hydrolyze GTP into GDP after binding with GTP, entering the inactivation state; this finally leads to the continuous activation of the downstream pathway, resulting in malignant proliferation, metastasis and anti-apoptosis of tumor cells [
60,
61]. Intrinsic GTPase and GTP-GDP exchange efficiency can differ between several mutant types of KRAS. For example, KRAS
G13 mutation is more sensitive to NF1-GAP-mediated hydrolytic activity, while KRAS
G12 and KRAS
Q61 mutations are insensitive to it [
62]. Another example is that the KRAS
G12C mutant type has similar intrinsic GTPase activity to the wild type, whereas other KRAS mutants have lower intrinsic GTPase activity than the wild type. [
20]. In fact, the KRAS
G12C inhibitor was designed with this characteristic in mind [
10].
It is also worth mentioning that the oncogenicity and drug resistance of mutant KRAS is related to its dimerization with wild-type KRAS [
63]. The exact relationship between them needs to be studied in depth.
1.2. Progress of PDAC with KRAS Mutations
The link between KRAS mutations and PDAC prognosis has been the focus of research, and several recent studies have further illustrated their relationship. Itonaga and colleagues analyzed the personal information of 110PDAC patients who underwent histological diagnosis from 2017 to 2019. All of these patients underwent first-line therapy with gemcitabine and nab-paclitaxel. Patients were analyzed for the presence of KRAS mutations and grouped through the quenching probe method. Then, progression-free survival (PFS) and overall survival (OS) were compared between the two groups. The study showed that patients with wild-type KRAS genes had much longer PFS and OS than patients with KRAS mutations (6.9/5.3 months (
p = 0.044) vs. 19.9/11.8 months (
p = 0.037), respectively) [
64]. In patients with surgically resectable tumors, KRAS gene mutations can also affect their prognosis after undergoing surgery. The analysis of patient data collected from Memorial Sloan Kettering (MSK) showed that patients with KRAS mutations had a worse prognosis after the surgical removal of the tumor [
65].
With the development of next-generation sequencing (NGS), it has become possible to measure the mutation frequency of the alleles in tumor samples [
66,
67]. As PDAC tumors are highly heterogeneous [
68], the proportion of malignant cells in tumors may vary greatly from patient to patient. Nauheim and colleagues studied microdissection samples from 144 PDAC patients who had undergone classic pancreaticoduodenectomy (PD) (classic Whipple) or pylorus-preserving PD (PPPD). KRAS mutations were present in 121 patients (84%). Studies show that patients with a high frequency of KRAS mutations (more than or equal to 20%,
n = 29) have larger tumors, higher postoperative distal recurrence rates, and shorter disease-free survival after surgery than those with a low frequency of KRAS mutations (less than 20%,
n = 29) [
69]. Another study found that PDAC patients who received FOLFIRINOX chemotherapy followed by the surgical resection of tumors had new KRAS mutations in their cell-free DNA compared to those before treatment [
70]. The relationship between increased KRAS mutations and chemotherapy, as well as the surgical resection of tumors, still warrants further exploration.
Research has progressed on the specific molecular mechanisms by which KRAS gene mutations worsen the prognosis of PDAC patients. It has been shown that KRAS
G12D, the most predominant KRAS mutant phenotype in PDAC, induces the overexpression of SUMO-activating enzyme subunit 1 (SAE1), which can lead to heterogeneous nuclear ribonucleoprotein A1 (hnRNPA1) being SUMOylated. SUMOylated hnRNPA1 is packaged by extracellular vesicles (EVs) and transported to human lymphatic endothelial cells (HLECs), ultimately promoting lymphatic vessel proliferation and lymph node metastasis [
11,
61].
2. KRAS Inhibitors for PDAC
2.1. KRASG12C Inhibitors
KRAS
G12C inhibitors have shown excellent results in the treatment of non-small cell lung cancer, and studies on their efficacy for other solid tumors are still advancing [
71]. A phase 1 trial (NCT03600883) evaluating the various aspects of sotorasib (AMG510) performance showed that sotorasib has good antitumor activity against solid tumors containing KRAS
G12C mutations [
49] (
Figure 2). Another KRAS
G12C inhibitor, MRTX849, validated its antitumor activity against KRAS
G12C mutation-containing tumors in a mouse xenograft model [
72]. However, none of the KRAS
G12C inhibitors have been approved by the FDA as a treatment for pancreatic cancer. Although the frequency of KRAS
G12C mutations in PDAC patients is abnormally high in some regions, for example, more than 60% in Japan [
73], the frequency of KRAS
G12C mutations in PDAC patients worldwide remains quite low, which leads to a limited prospect for the clinical treatment of PDAC using KRAS
G12C inhibitors [
11,
74].
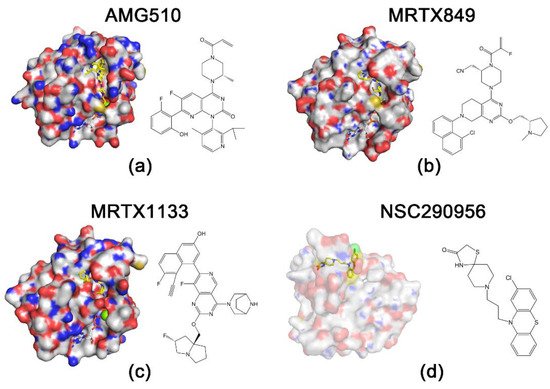
Figure 2. Structures of RAS proteins and inhibitors. Protein is indicated by surface representation, and compounds and nucleotides are shown in stick models. The carbon and hydrogen atoms of the inhibitor are marked in yellow to highlight them. (
a) KRAS
G12C and AMG510 (Protein Data Bank (PDB): 6OIM). (
b) KRAS
G12C and MRTX849 (PDB: 6UT0). (
c) KRAS
G12D and MRTX1133 (PDB: 7RPZ). (
d) HRAS
G60A and NSC290956 [
14].
2.2. KRASG12D Inhibitors
2.2.1. MRTX1133
While sotorasib has been approved by the FDA for the treatment of KRAS
G12C mutation-containing NSCLC [
51], the development of other KRAS mutation inhibitors has come to a standstill. One of the main reasons hindering the development of KRAS
G12D inhibitors, which has been mentioned previously, is the low rate of intrinsic GTP hydrolysis in the KRAS
G12D mutant [
60]. KRAS mutations lead to a decrease in intrinsic GTPase activity, which further decreases the rate of GTP hydrolysis and ultimately continues to activate downstream pathways and produce carcinogenesis [
61]. The intrinsic hydrolysis rate of the KRAS
G12C mutation is equivalent to approximately 70% of that of the wild-type KRAS, while the intrinsic hydrolysis rate of the KRAS
G12D mutation is only less than 30% [
60]. This disadvantage poses a challenge for the design of KRAS
G12D inhibitors. It is also challenging to determine whether the inhibitor has sufficient affinity for 12-aspartate involved in the KRAS
G12D mutant to avoid binding to wild-type KRAS. In February 2022, Mirati Therapeutics announced a selective non-covalent inhibitor, MRTX1133 of KRAS
G12D (
Figure 2). The structure of MRTX1133 is based on MRTX849, a KRAS
G12C inhibitor developed by Mirati Therapeutics. The investigators introduced a salt bridge between the inhibitor and 12-aspartate to enhance the reversible affinity for KRAS
G12D. This strengthened the selectivity of the inhibitor for KRAS
G12D through a series of modifications to avoid binding to wild-type KRAS. Compared to several KRAS
G12C inhibitors whose reversible affinity for the target is in the micromolar range [
50,
75,
76], MRTX1133 has a picomolar range of reversible affinity for KRAS
G12D. Although MTRX1133 binds weakly to KRAS proteins in the GDP state, it also has the ability to bind to KRAS proteins in the GTP state [
77]. This will lead to new ideas for combination therapy studies of KRAS inhibitors. In a previous study, MRTX1133 achieved excellent results in a mouse xenograft model of pancreatic cancer, with a 94% reduction in tumor volume at 3 mg/kg BID (IP) compared to the control group [
13].
2.2.2. Peptide Nucleic Acids (PNAs)
Peptide Nucleic Acids (PNAs) are synthetic nucleotide analogs whose molecular structures are very similar to those of DNA and RNA [
78]. PNAs have good hybridization properties and can specifically bind to complementary DNA or RNA, distinguishing similar sequences even at the level of single base mismatches [
79,
80]. Meanwhile, PNAs can bind specifically to the mRNA of the target gene and inhibit its translation process [
81]. Moreover, PNAs have stable chemical structures and are not easily degraded by nucleases or proteases. Based on the above characteristics, treatment using PNAs has great potential to become a new tool in the fight against malignant tumors. In a recent study, several PNAs were designed for the KRAS
G12D mutated gene fragment and tested in the human metastatic pancreatic adenocarcinoma cell line AsPC-1 containing the KRAS
G12D mutation. The results showed that PNAs significantly inhibited tumor cell activity and reduced the expression of the KRAS
G12D mutated gene [
82]. The successful inhibition of the KRAS
G12D mutant gene by PNAs at the cellular level raises the possibility for subsequent animal experiments.
2.3. Pan-RAS Inhibitors
Compared to specific inhibitors, pan-RAS inhibitors have broader applicability and can provide treatment for patients with different types of KRAS mutations. Additionally, pan-RAS inhibitors can avoid drug resistance caused by the compensatory activation of wild-type KRAS. Although this class of inhibitors suffers from high toxicity and off-target inhibition, it still has great research potential [
83]. Several pan-RAS inhibitors have been shown to have good specificity for RAS mutations, and animal models have tolerated these inhibitors to an appreciable degree [
84,
85].
Nassar et al. revealed that there are three distinct but equally populated conformations in the process of HRAS-GTP hydrolysis and nucleotide exchange, one of which is the “non-signaling open conformation” state [
86]. Due to the same hydrolysis process and the structural homology, the state also appears in KRAS [
87]. Using nuclear magnetic resonance (NMR) analysis, the researchers uncovered that the HRAS
G60A-GppNp complex adopts an “open conformation” at the switch 1 region and abolishes the biological activity of HRAS [
86,
88]. Recent studies have indicated extremely open switch 1 conformations of KRAS [
89]. This implies that the “open conformation” may be a convergent point for survival signaling in KRAS-driven cancer, and agents locking this “open conformation” may theoretically block KRAS-dependent signaling. Most recently, Jin Wang’s group used a Specificity Affinity (SPA)-based virtual screening strategy to develop small-molecule inhibitors that stabilize the “open conformation”. This process led to the selection of three hits (NSC290956, NSC48693, and NSC48160) from 2000 compounds by individually docking compounds in the National Cancer Institute diversity compound sets to the “open non-signaling intermediate conformation” of RAS [
89]. Of these, NSC290956 (also termed Spiclomazine or APY606) manifested potent efficacy against the proliferation of KRAS-driven pancreatic cancer cell lines CFPAC-1 (KRAS
G12V), MIA PaCa-2 (KRAS
G12C), Capan-1 (KRAS
G12V), SW1990 (KRAS
G12T) and BxPC-3 (wild-type KRAS) and pancreatic cancer cells but showed much less toxicity towards human normal cells [
15,
90,
91]. NSC48160 inhibited the survival and growth of KRAS-driven pancreatic cancer cells CPFAC-1 (KRAS
G12V) and BxPC-3 (wild-type KRAS) by using MTT and colony-forming assays [
16]. Liu et al. found that NSC48160 selectively induced apoptosis in pancreatic cancer MIA PaCa-2 (KRAS
G12C) cells as compared to human normal HEK-293 and HL-7702 cells [
92]. Liu et al. further found that the inhibitory effects of small-molecule NSC48693 on KRAS-driven cancer cells were greater than NSC48160 for CFPAC-1(KRAS
G12V), MIA PaCa-2 (KRAS
G12C) and BxPC-3 (wild-type KRAS) cells [
93]. Interestingly, the cytotoxic effect of NSC48693 on the human normal cell line (HL-7702) was lower than that on cancer cell lines (CFPAC-1, MIA PaCa-2 and BxPC-3). Together, this research provides functional insights into the “open conformation” and validates three hits acting as pan-KRAS inhibitors to induce the apoptosis of pancreatic cancer cells.
This entry is adapted from the peer-reviewed paper 10.3390/cancers14204982