Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Andrea Catarina Amaro de Campos Lobo | -- | 4211 | 2022-10-19 10:40:53 | | | |
| 2 | Rita Xu | Meta information modification | 4211 | 2022-10-20 08:21:05 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Amado, B.; Melo, L.; Pinto, R.; Lobo, A.; Barros, P.; Gomes, J.R. Ischemic Stroke Pathophysiology and Preclinical Models. Encyclopedia. Available online: https://encyclopedia.pub/entry/30161 (accessed on 23 July 2026).
Amado B, Melo L, Pinto R, Lobo A, Barros P, Gomes JR. Ischemic Stroke Pathophysiology and Preclinical Models. Encyclopedia. Available at: https://encyclopedia.pub/entry/30161. Accessed July 23, 2026.
Amado, Beatriz, Lúcia Melo, Raquel Pinto, Andrea Lobo, Pedro Barros, João R. Gomes. "Ischemic Stroke Pathophysiology and Preclinical Models" Encyclopedia, https://encyclopedia.pub/entry/30161 (accessed July 23, 2026).
Amado, B., Melo, L., Pinto, R., Lobo, A., Barros, P., & Gomes, J.R. (2022, October 19). Ischemic Stroke Pathophysiology and Preclinical Models. In Encyclopedia. https://encyclopedia.pub/entry/30161
Amado, Beatriz, et al. "Ischemic Stroke Pathophysiology and Preclinical Models." Encyclopedia. Web. 19 October, 2022.
Copy Citation
Ischemic stroke is a leading cause of death worldwide, mainly in western countries. So far, approved therapies rely mainly on reperfusion of the affected brain area, by intravenous thrombolysis or mechanical thrombectomy. The combination of pharmacological brain-protective strategies with reperfusion is the future of stroke therapy, aiming to reduce brain cell death and decrease patients’ disabilities. The success of new therapies relies on bringing preclinical studies and clinical practice close together. Recent upgrades of in vitro and in vivo stroke models for accurate and effective evaluation of therapeutic strategies are described.
ischemic stroke
cytoprotection
spheroids
organoids
MCAO
preclinical
therapies
1. Stroke
Stroke was described by the World Health Organization (WHO) in 1970 as a syndrome with “rapidly developing clinical signs of focal or global disturbance of cerebral function, lasting more than 24 h or leading to death, with no apparent cause other than of vascular origin” [1]. In recent years, the American Heart Association (AHA) and the American Stroke Association (ASA) consider this definition outdated, because it is mainly focused on clinical symptoms [2] and does not reflect significant advances in the “nature, timing, clinical recognition of stroke and its mimics, and imaging findings” [2]. As a result, in 2013, the AHA and ASA revised the stroke definition to include silent infarctions (cerebral, spinal, and retinal) and silent hemorrhages [1][2][3], and highlighted that stroke is both a clinical and a radiological diagnosis [1]. Nowadays, ischemic stroke is defined as a focal neurological deficit triggered by a vascular cause and traced to the central nervous system (CNS) [4]. The cessation of blood flow to the brain leads to an absence of oxygen and nutrients, causing extensive cell damage and neuronal death [5].
Stroke is the world’s second most common cause of death, and one of the major causes of adult disability and dementia [1][4][6][7]. Stroke events affect 13.7 million individuals per year worldwide, causing the death of around 5.5 million people each year, and constitute a huge global health burden [1][4][6][8]. Ischemic strokes account for about 71% of all strokes [9], and are caused by a reduction in blood flow caused by the occlusion of a brain artery [4][6][8]. The remaining 29% of stroke events are hemorrhagic originated by a cerebral artery rupture, and causing internal bleeding [4][6].
During the 1990–2016 period, the prevalence of stroke in low and middle-income countries doubled, while it decreased by 42% in high-income countries [10]. In 2017, the predicted cost for stroke management was €45 billion—including direct and indirect costs [11]. As reported by the Global Cost of Disease Study (GDB), the stroke survival rates improved, but the socio-economic burden increased, since treatments available reduced stroke mortality and increased the number of disabled [10][12]. A study estimates that by 2035, the number of strokes, globally, will increase by 34%, as populations grow and people live longer, with the number of survivors increasing by 25% [12]. Thus, it is essential to establish better prevention and management plans for stroke, especially recovery systems.
Primordial prevention of stroke focuses on educating the population [13], through educational campaigns, public lectures, and information stalls, to raise awareness of risk factors, healthy lifestyles, and stroke symptoms [12]. Although prevention of stroke also carries costs, it contributes to decreasing stroke rates [12].
2. Ischemic Stroke
Ischemic strokes are caused by thrombotic or embolic events that lead to the reduction of blood and oxygen supply to the brain [14][15]. The cause of the stroke has an impact on both prognosis and outcomes [14][15]: thrombosis, the most common, happens when a clot is formed in a vessel of the brain or neck [16]; embolism occurs when a clot moves from another part of the body to the brain [16].
Ischemic strokes can be classified as acute (AIS) or transient ischemic attacks (TIAs). AIS requires rapid and effective treatment to limit irreversible damage and persistent symptoms, TIA usually resolves in less than 24 h, by spontaneous reperfusion, and does not lead to significant damage or symptoms [16].
2.1. Risk Factors
More than 90% of the stroke burden is caused by modifiable risk factors [16]. An event of stroke can occur to any person, of any sex, age, ethnicity, or social class [17], but some individuals are more prone to stroke considering risk factors identified by epidemiological studies [18].
Non-modifiable risk factors include age, sex, race, and genetic factors [19]. Studies report that after 55 years of age, the risk of suffering a stroke event nearly doubles for each subsequent decade [18]. Regarding sex, the incidence in men is 33% higher than in women, although the severity is greater in women [20]. Concerning race/ethnicity, a study reports that in the USA, black individuals (both men and women) between 45 and 84 years old are three times more likely to suffer a stroke event than white individuals; this difference scatters beyond 85 years old [21]. Ischemic strokes can also be caused by highly penetrant Mendelian mutations [22][23], meaning that familiar history of stroke raises the probability that an individual will suffer a stroke [22][23]. Some of these loci are well-known [24][25], so patients carrying those mutations can be aware of the risk. Genetically caused strokes are typically early in onset and lack the usual risk factors [24][25]. Many of these non-modifiable risk factors harm the cardiovascular system, which is frequently the strongest indicator to assess stroke risk [26]. Hence, as people get older, atherosclerosis worsens, increasing their risk of suffering an ischemic stroke and myocardial infarction [27].
Modifiable risk factors include smoking, obesity, drinking alcohol, high-stress levels or anxiety, and a sedentary lifestyle [19]. Underlying disorders, such as hypertension, Diabetes Mellitus, heart/blood vessel diseases, or brain aneurysms [19] also take a role as stroke risk factors and should be monitored.
The single most important risk factor is hypertension [4]. In the United States, 46% of adults have high blood pressure [28], and in Europe, this risk factor is under-treated, with studies revealing disease control rates of 32% or less in people with established hypertension [12]. The WHO states that for every 10 stroke-caused deaths, four could be prevented if blood pressure was regulated [29]. A poor diet is the second leading factor related to stroke mortality, with diabetes and obesity accounting for the third and fourth most important risk factors, respectively [29]. Smoking is the fifth biggest risk factor—smokers display 4.5 times increased stroke mortality [29].
In recent years, significant advances in prevention plans and management of vascular risk factors, such as smoking cessation, and control of hypertension, hypercholesterolemia, and diabetes mellitus contributed to a decrease in stroke incidence [30]. Physical exercise reduces cardiovascular and cerebrovascular risk and boosts the expression of neuroprotective factors, effective to prevent stroke [26]. Concluding, it is critical to promote and support healthy behaviors, such as weight loss, a balanced diet, and frequent exercise [26][30].
2.2. Diagnosis
The most common signs of ischemic stroke are sudden numbness/weakness on one side of the body (hemiparesis) [4], confusion and difficulty speaking, trouble seeing with one or both eyes, sudden dizziness/loss of coordination, and acute headache with unknown cause [16][31]. Less common symptoms include nausea/vomiting, vertigo, and loss of conscience [4]. Stroke symptoms can be sudden (the most common), or develop slowly and progressively [32]. Time management of the signs and symptoms is extremely important, referred to as the time of onset (the time when signs first occurred, reported by the patient or observers) [33].
The patient’s prognosis may be improved if the ischemic stroke is diagnosed early [34]. Fortunately, the awareness of stroke’s signs and symptoms was popularized by some common methods. The face arm speech test (FAST), a rapid procedure intended to assist ambulance staff to recognize acute stroke, promotes early stroke detection and diagnosis [34]. A focused neurologic examination, ideally incorporating the National Institute of Health Stroke Scale (NIHSS), should be performed to determine stroke severity, recording the heart rate and rhythm, blood pressure, and the presence or absence of fever [30].
Following the first approach, imaging procedures should confirm stroke diagnosis, and distinguish the subtype of stroke, aiding with possible treatments [30][34]. In most institutes, non-contrast head computed tomography (NCT) is the first course of study [30][34]. This is a less expensive, quicker, and commonly available technique, compared to magnetic resonance imaging (MRI), but the extent and severity of the perfusion deficit are not provided by NCT [34]. Early MRI is useful to confirm stroke diagnosis, the time of onset, and the infarcted core [30]. Both ER and imaging services must be prepared for rapid screening, which is critical to define the appropriate treatment to save as much brain tissue as possible, allowing a better prognosis.
AIS management has changed drastically in recent years, with more individuals obtaining treatment to reduce death and long-term disability [35][36]. A key breakthrough was to create coordinated regional stroke care systems to promptly identify stroke patients and direct them to reference centers offering appropriate therapy [35][37][38]. This entails performing clinical and imaging evaluations by clinicians to identify the patients that are eligible for treatment [35][37][38]. Thus, stroke networks and protocols in healthcare systems are critical to ensure that patients receive prompt treatment.
2.3. Key Concept: Core and Penumbra
When an ischemic stroke occurs, blood flow and oxygen supply to the brain diminish rapidly [19] to a critical level [4]. In AIS, the parenchymal lesions can be divided into two morphological zones: the ischemic core and the penumbra [39][40]. The almost complete lack of perfusion defines the core area, characterized by dead or dying tissue in the center of the infarct—this site is irreversibly damaged [16][19]. In this region, the reduction of blood flow below 15–20% of regular levels causes neuronal necrotic death [16] that cannot be stopped by neuroprotective drugs [39]. Core size depends on occlusion rate, collateral blood flow, and how quickly reperfusion is achieved [4][16][41]. The ischemic core growth is closely related to a worse prognosis for patients [4].
The penumbra includes brain tissue that is partially perfused and degenerates more slowly [39][40]. This area is a dynamic zone, potentially salvageable, that displays loss of neuronal function but maintains cellular integrity [9][16]. The penumbra zone is subdivided into an apoptotic area (potentially salvageable) and live cells [42]. However, if not promptly reperfused, the penumbra will be incorporated into the ischemic core (Figure 1) [39][40]. To guide stroke therapy, it is necessary to estimate accurately the penumbra size, through modern imaging techniques, and to treat patients as soon as possible to reduce significantly penumbra extent, and prevent core growth [39][40].
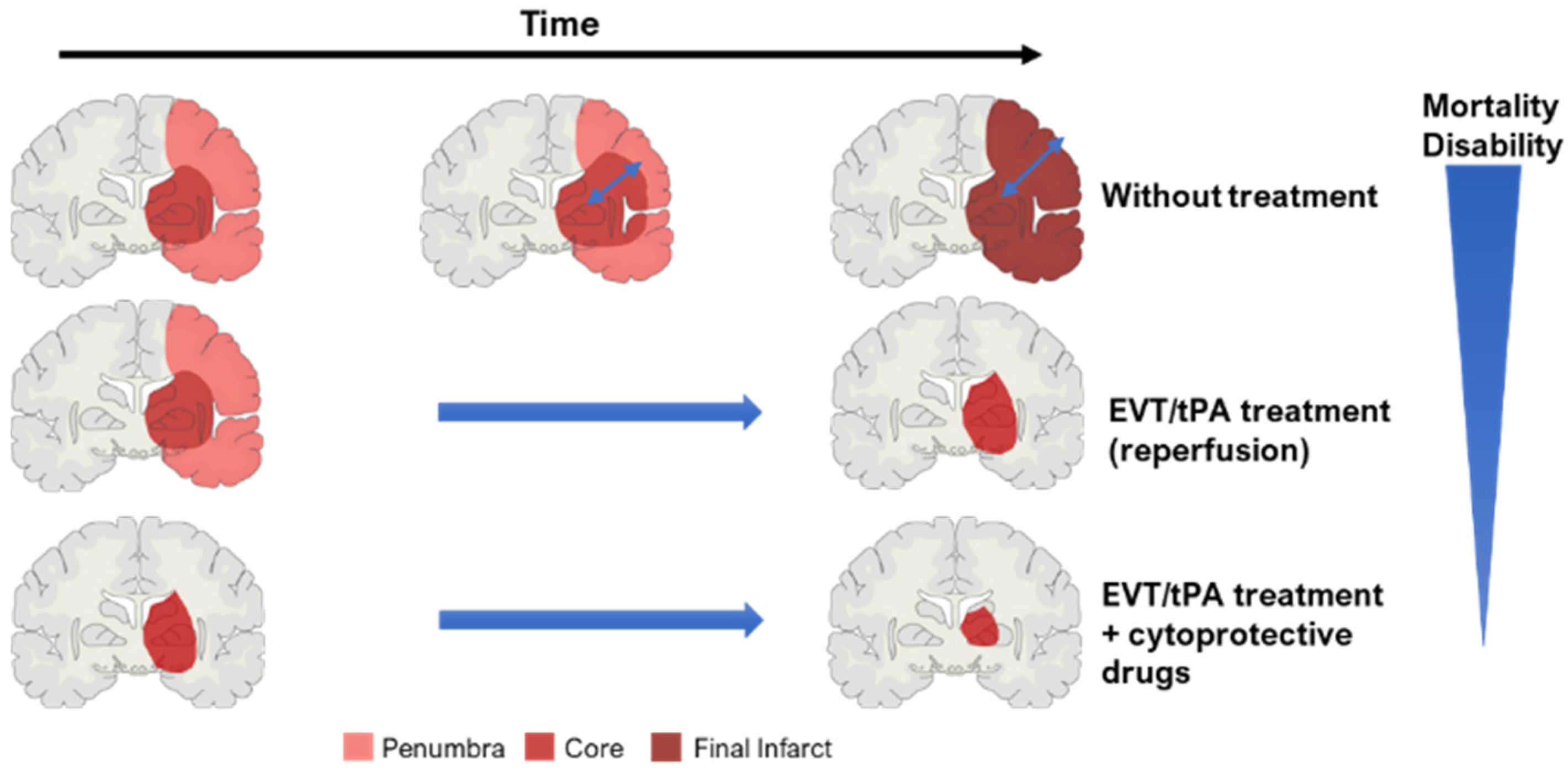
Figure 1. Concept of ischemic core and penumbra, and their progression through time (EVT—endovascular/mechanical thrombectomy; tPA—thrombolysis). The blue arrow within the brain in the ‘without treatment’ condition represents core expansion.
2.4. Ischemic Stroke Pathophysiology
Although occlusion time determines the size of the injury and stroke outcomes, several pathways are triggered, even if reperfusion is achieved quickly [42]. Stroke’s physiological mechanisms are very complex and intertwined, with excitotoxicity, oxidative stress, and inflammatory processes playing a major role in neuronal damage [43].
The brain receives 20% of cardiac output at rest and is extremely vulnerable to ischemia. Even brief ischemic episodes can set off a complicated chain of events that can lead to lifelong cerebral damage [44]. Critically lowered cerebral blood flow (CBF) during brain ischemia, results in insufficient oxygen and glucose delivery, beginning the stroke pathogenic process (Figure 2) [44][45]. The lack of oxygen and nutrients impairs cellular metabolism and leads to a reduction in energy production, with consequent failure of energy-dependent systems, such as the sodium-potassium ATPase [44][46]. This ionic pump is crucial to maintain the ionic gradients across the neuronal membrane, meaning that when not working properly, the ionic imbalance leads to an increase in the release/inhibits the reuptake of excitatory neurotransmitters, such as glutamate [46]. Prolonged exposure to glutamate overstimulates ionotropic N-methyl-D-aspartic acid (NMDA) and 1-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptors, causing an increase in calcium influx and leading to downstream activation of enzymes that breakdown cellular membranes, proteins, nucleic acids, and contribute to a buildup in oxidative stress [44][46]. These processes can culminate in neuronal death.
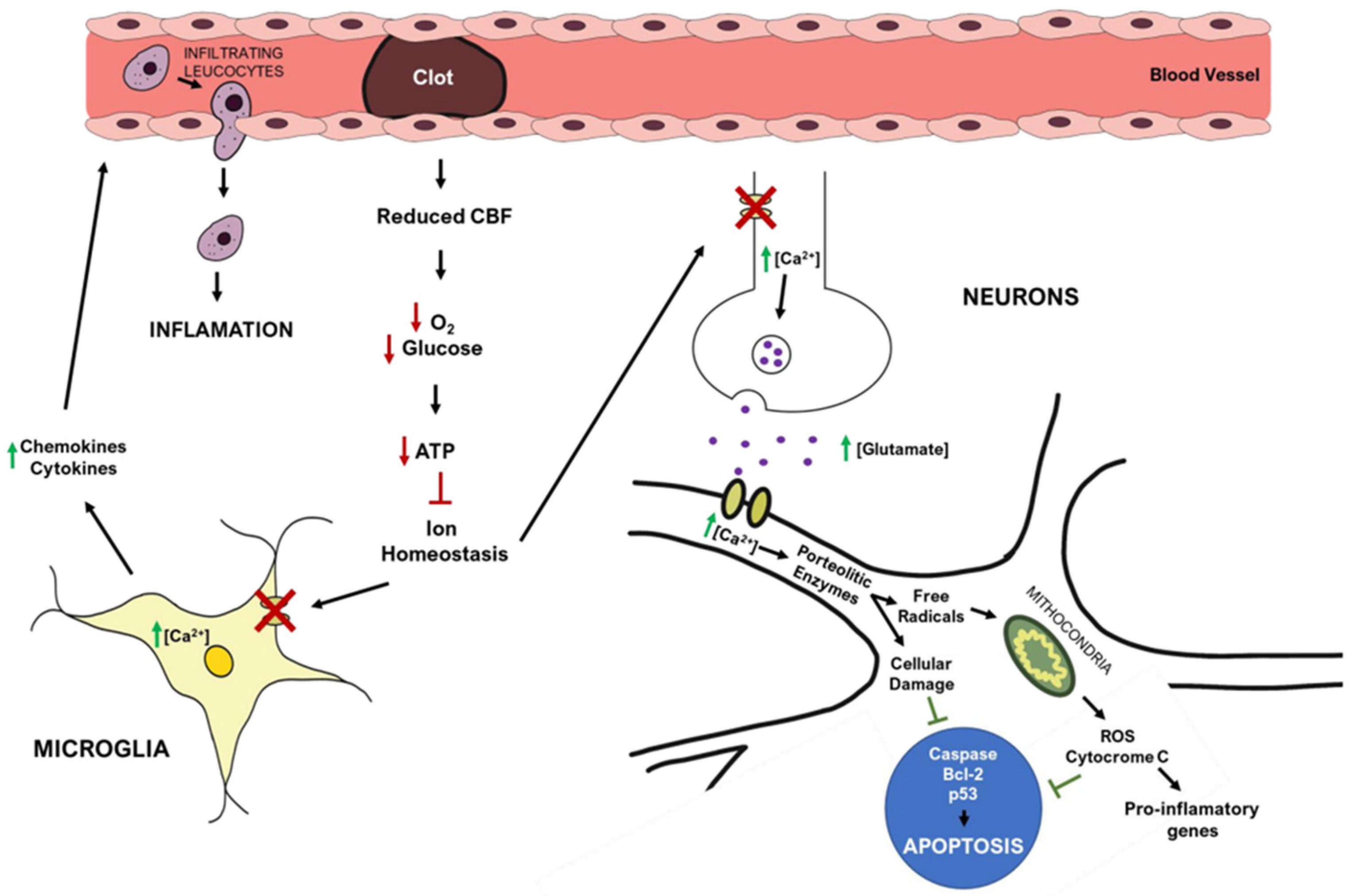
Figure 2. Simplified overview of stroke’s pathophysiology: molecular pathways occurring in the brain after ischemic stroke. Red arrows represent decreased events, while green arrows represent increased events.
The mitochondria play a central role in metabolism, being responsible for energy production (ATP) via the electron transport chain, a process that also yields the release of reactive oxygen species (ROS). After ischemia, high levels of intracellular Ca2+, Na+, and adenosine diphosphate (ADP) induce excessive mitochondrial oxygen radical production [46], overwhelming endogenous scavenging mechanisms and causing direct damage to lipids, proteins, nucleic acids, and carbohydrates [44][45][46]. In particular, oxygen radicals and oxidative stress promote the formation of mitochondrial transition pores, compromising mitochondrial integrity, and leading to cytochrome c release (a trigger of apoptosis) [44][46]. Ischemia also increases the expression of pro-apoptotic Bc-l2 family members and p53 genes [24], activating caspases and calpain cascades that cause cellular apoptosis [24].
Post-ischemic inflammation is also a relevant process in stroke pathophysiology, with contributions from endothelial cells, astrocytes, microglia, and neurons [44][45][46]. Increased Ca2+, ROS production, and the lack of oxygen and nutrients can activate astrocytes and microglia. These cells produce pro-inflammatory cytokines, such as interleukin-1 (IL-1), tumor necrosis factor-1 (TNF-α), and interleukin-1β (IL-1β) [44][45][46], that reduce cell adhesion molecules and impair the function of the extracellular matrix. These events are critical to the infiltration of inflammatory cells into the brain parenchyma and for increasing brain inflammation [44][45].
Apoptosis is triggered by the generation of toxic mediators, produced by the activated inflammatory cells and damaged neurons [24].
However, the ischemic cascade also activates neuroprotective mechanisms that might limit apoptotic and necrotic cell death or display anti-inflammatory properties [24], such as increased expression of heat shock protein 70 (HSP70), Bcl-2 anti-apoptotic proteins, prion protein (PrP), neurotrophin-3 (NT-3), and the anti-inflammatory interleukin-10 (IL-10) [24].
2.5. Treatments
Currently, reperfusion of the ischemic brain remains the only available treatment. It consists in the restoration of the blood circulation and re-oxygenation of the affected tissues [47]. This treatment was introduced in 1995 [48], and despite occasionally having a contradictory effect (causing increased damage to the tissues) [47], it remains the most effective treatment for acute ischemic stroke [49][50]. The typical reperfusion technique provides a short time window for treatment, with only a small percentage of patients (usually 1% to 3%) receiving reperfusion therapy, due to a sizable risk of symptomatic hemorrhagic transformation [49].
Reperfusion therapies save penumbral tissue, reduce final infarct size, and enhance clinical outcomes, by restoring blood flow to vulnerable tissues before they advance into infarction [49][50]. The first approved reperfusion treatments consisted of thrombolytic methods—intravenous (IV) tissue plasminogen activator (tPA) [50]. Only in 2015, after years of research and clinical trials [51], endovascular techniques were approved as a reperfusion method—relying on methods such as mechanical thrombectomy (MT) [50].
2.5.1. Thrombolysis
Thrombolysis is a pharmacological treatment that involves the infusion of tissue plasminogen activator—tPA, or a tPA analog—to promote the lysis of the blood clot via the activation of the proteolytic enzyme plasminogen into plasmin [50]. Plasmin cleaves cross-link connections between fibrin molecules, which provide the structural scaffold for blood clots [50]. Clots became soluble and are further degraded by other enzymes, allowing blood flow to be restored [50]. The three principal types of plasminogen activators include tPA, streptokinase (SK), and urokinase (UK), and they differ in the mechanism of action on fibrin molecules [50]. Alteplase (recombinant tPA—rtPA), Retaplase, and Tenecteplase (TNK-tPA) are examples of recognized tPA analogues [50].
In 1995, a National Institute of Neurological Disorders and Stroke clinical trial changed AIS treatment, by demonstrating the safety and efficacy of intravenous (IV)-tPA [51][52]. It demonstrated that when IV-tPA is administered up to 3 h after symptom onset, patients are around 30% more likely to have only minor to no disability at 90 days following stroke [51][52]. The limited time window for IV-tPA usage in ischemic strokes makes it available to only 3.2–5.2% of AIS patients in the USA [51][52]. The AHA/ASA amplified the IV-tPA window from 3 to 4.5 h in 2009, based on European research, increasing the usage of this treatment to 20% of the patients [52].
Thus far, alteplase is the only thrombolytic drug approved by the Food and Drug Administration (FDA) for the treatment of AIS [50]. Recent advances were made with other thrombolytic drugs, such as tenecteplase or desmoplase, both in [41] Phase III trials (in 2020 and 2018, respectively). These therapies display several advantages over alteplase: [41] tenecteplase shows a reduced risk of hemorrhagic transformation [41]; desmoplase can be safely administered up to 9h after symptoms onset [41]. Furthermore, a recent trial (EXTEND-IA TNK) demonstrated that tenecteplase leads to better functional outcomes than alteplase [53]. Tenecteplase was introduced to stroke guidelines as an alternative to alteplase after these findings, and some stroke units use it frequently as a substitute for alteplase [54][55].
2.5.2. Mechanical Thrombectomy
Mechanical thrombectomy (MT) is another extensively used treatment, after the publication in 2015 of five clinical trials showing benefits in MT up to 6 h after stroke symptoms onset [51][52]. Multiple trials have indicated that MT, in addition to normal medical therapy, improves the overall outcome of AIS patients with occlusion of the proximal middle cerebral artery (MCA) or internal carotid artery (ICA), when treated until 24 h after symptom onset [51][52]. Thanks to MT, the temporal frame for AIS treatment has been extended, providing clinicians with more robust therapeutic armament [52]. Modern MT more than doubles the probability of a better functional outcome compared to standard therapy alone, according to a pooled meta-analysis, with no significant difference in mortality or risk of parenchymal hemorrhage after 90 days [52].
Instead of breaking down the clot with chemicals, microcatheter devices (stents) are used in mechanical thrombectomy to remove the blood clot from an occluded artery with minimally invasive surgery [50][51]. This technique provides a larger time window than thrombolysis, with fewer adverse effects [51][56]. Additionally, when stents cannot access some brain areas or the clot is not easily removed, the aspiration technique might be used, [56] to aspirate the clot with a catheter and retrieve it [56].
Recent developments in the field of MT consist of combining stent retriever techniques with aspiration [57], increasing the rates of successful reperfusions. Furthermore, the establishment of guidelines for anesthesiology (as presented in the GASS trial [58]) and post-thrombectomy management [57] are important factors to increase the success of this technique. The Mechanical Thrombectomy with or without Intravenous Alteplase in Acute Stroke trial is the first study that discusses the hypothesis that mechanical thrombectomy alone might not be inferior to the combination of mechanical thrombectomy with intravenous alteplase, in patients eligible for both treatments. Previously, several observational cohort studies have investigated the outcome of intravenous thrombolysis before mechanical thrombectomy [59][60][61]. A study comprising 656 patients in China shows that mechanical thrombectomy alone within 4.5h of stroke onset is similar to mechanical thrombectomy preceded by intravenous alteplase, in a three-month functional follow-up [59][62].
Concluding, thrombolysis and mechanical thrombectomy are used to stop ischemia and restore the brain’s blood supply, but they do not limit the brain damage caused by inflammation and other processes during reperfusion [51]. Thus, pharmacological therapies that limit cell damage are required to improve stroke treatments.
2.5.3. Neuroprotective Drugs
The primary purpose of therapeutic intervention in stroke is to reduce lethality and comorbidities associated, leading to good neurological outcomes for the survivors—in terms of motor coordination, memory, and speech [63]. However, even with successful reperfusion, not all patients recover their motor and neurological function [4]. Therefore, the development of novel neuroprotective therapies is a growing research field.
There is a massive effort from the scientific community to find pharmacological therapies that protect the cerebral tissue after brain ischemia [64]. For decades, the research focused on neuroprotection, considering that neurons were the main affected cells by ischemia [63][65]. Nowadays, it is considered that glial cells also take part in ischemic stroke pathophysiology [65], mainly in the inflammatory response. Hence, a more comprehensive approach to the development of brain protective drugs was established, including the study of oligodendrocytes, astrocytes, and microglial cells, as well as neurons [66].
The recent concept of the neurovascular unit (NVU), which integrates glial cells, neurons, vascular cells, and the basal matrix, was a major milestone in brain-protective research [67]. The focus is to develop therapies that mitigate the damage after stroke and diminish/ reverse apoptosis in the penumbra area, impacting all cell types comprised in the NVU [68]. Several studies indicate that reperfusion therapies combined with cytoprotective drugs can extend the therapeutic window, and reduce the damage to the blood–brain barrier (BBB) and the risk of hemorrhagic transformation post-ischemia [38][63]. Moreover, the recent discovery of diverse patterns for cell death and reperfusion is opening the doors for discoveries regarding cytoprotectants [69]. Cytoprotective drugs are now complementary to reperfusion, and not administered alone, which is a new step for successful therapies [70].
Due to the complexity of the events occurring during stroke, several types of protective drugs were investigated (Table 1). The first type centered on the blockage of voltage-gated cation channels [63], especially calcium channels [63][68]. Several drugs targeting this mechanism were tested. Nimodipine, one of the first candidates to go through clinical trials [71], blocks L-type calcium channels, preventing calcium influx and vasoconstriction [72]. The VENUS trial, a phase III clinical trial, tested nimodipine 6 hr after stroke onset [73], but was terminated prematurely since it was not effective [71]. A small Phase I clinical trial (MAVARIC) is investigating the efficacy and safety of verapamil (a calcium channel blocker) and magnesium administration after reperfusion [74]. The results are yet to be published, but another trial demonstrated that magnesium sulfate did not improve functional outcomes, when administrated alone [75].
Table 1. Overview of recent clinical trials for cytoprotective therapies—phase, status, and mechanism of action.
| Clinical Trial | Therapeutic Drug | Mechanism of Action | Status of Clinical Trial |
|---|---|---|---|
| VENUS (Phase III) | Nimodipine | Blocks voltage-gated channels (calcium) | Terminated: not effective |
| MAVARIC (Phase I) | Verapamil (with magnesium sulfate) | Blocks of voltage-gated channels (calcium), after reperfusion | Results not published yet |
| ESCAPE-NA1 (Phase III) | Nerinetide (Na-1) | Inhibits neuronal excitotoxicity | Approved by American FDA alone; but it does not display protection together with IVT |
| ESCAPE-NEXT (Phase III) | Nerinetide (Na-1) together with EVT therapy | Inhibits neuronal excitotoxicity | Ongoing (completes in August 2023) |
| URICO-ICTUS (Phase III) | Uric Acid (UA) | Antioxidant agent (reduces inflammation) | Small study sample, but a confirmatory trial is planned |
| BAST (Phase III) | 3-N-butylphtalide (NBP) | Free radical scavenger (reduces inflammation) | Ongoing (completed on 31 December 2022), good outcome so far |
| Evaluation of HUK in AIS (Phase IV) | Human Urinary Kallidinogenase (HUK) | Anti-inflammatory agents suppress TLR4/NF-kB pathway | Approved by China’s FDA, with post-approval surveillance ongoing |
| A study of allogeneic mesenchymal bone marrow cells in AIS (Phase I/II) | Allogeneic adult mesenchymal bone marrow cells | Cell-based therapy | Completed, presented beneficial behavioral outcomes for patients |
| J-REPAIR (Phase I/II) | JTR-161 (allogeneic stem cell product) | Cell-based therapy | Last updated on June 2022, results not published yet |
| Efficacy and safety of FTY720 for AIS (phase II) | Fingolimod (FTY720) | Immune modulator | Completed; reduced injury and improved clinical outcomes |
| Combination of the immune modulator Dimethyl Fumarate with alteplase in AIS (phase I/II) | Dimethyl Fumarate | Immune modulator | Ongoing (completes in December 2022) |
Therapies targeting glutamate excitotoxicity, one of the main processes in stroke pathophysiology, were also tested. An example is N-methyl-D-aspartic acid receptor (NMDAR) antagonists. NMDARs are glutamate receptors, which are overactivated during excitotoxicity [63][68] and contribute to neuronal death [49]. The inhibition of these receptors using, for instance, MK801, reduces the neuronal response to glutamate and brain infarct; in animal models, MK801 reduced infarct volume by up to 75% [68]. However, this drug blocks the whole excitatory pathway, causing side effects such as memory loss [68], and has a short therapeutic window, which stopped its progression to clinical practice [76]. Nerinetide (aka. NA-1), a therapy targeting excitotoxic mechanisms was developed recently. The post-synaptic density protein 95 (PSD-95) is a scaffolding molecule [77] that binds NMDAR, triggering different intracellular pathways [78]. In excitotoxic conditions, such as the ones occurring in stroke, PSD95-NMDARs interaction causes Ca2+ overflow, leading to increased production of nitric oxide (NO), and activating pro-apoptotic factors [78]. Thus, NA-1 interferes with the PSD-95 [72] interaction with NDMARs and suppresses this excitotoxic pathway [79]. In macaque models of MCA occlusion, this drug was demonstrated to be benefic, reducing infarct volume, so it advanced to a Phase III clinical trial. ESCAPE-NA1 trial proved that nerinetide reduced mortality, improved clinical outcomes, and reduced infarct volume [80]—based on these outcomes, this therapy was approved by the FDA [81]. However, in patients receiving intravenous thrombolytic agents therapy, nerinetide was not effective [80], since tPA produces plasmin that cleaves and inactivates nerinetide [81]. A new trial, ESCAPE-NEXT, aims to combine nerinetide and mechanical thrombectomy for patients with acute ischemic stroke [82]. This trial is expected to be completed in August 2023, with outcomes assessed 90 days after the drug administration [82].
Another therapeutic target to prevent stroke-induced cell damage is the suppression of apoptosis [71], by inhibiting apoptotic proteins from several pathways. Despite conferring cell protection, the effects were transient and cell death occurred afterward [42]. Some researchers believe that molecules from a higher level of regulation should be considered: for example, upregulation of the angiotensin II type 2 receptor increases the expression of brain-derived neurotrophic factor, which can reduce caspase-3 activity, and consequently apoptosis [83]. Despite this, no apoptotic inhibitor has reached clinical trials, since the mechanism of action is irreversible, and has poor brain penetration [71].
Free radicals production is a major cause of stroke damage [68]. Free radicals are unpaired electron molecules, such as ROS, that cause cell death due to their instability and reactivity [63]. The use of free radical scavengers and antioxidants may protect the brain cells after a stroke [68]. NADPH-oxidase (NOX) inhibitors, as well as other free radical scavengers, interact with ROS, converting them into stable molecules [68][84]. Uric acid acts as an antioxidant agent and reduces brain swelling, inflammation, and apoptotic death [85]. Despite the uric acid mechanism of action is not fully understood [85], it evolved into Phase III clinical trial (URICO-ICTUS) [85], where it improved patients’ functional outcomes. Nonetheless, the sample size was restricted (421 patients) [86], and a confirmatory trial to further test uric acid as a potential stroke therapy is planned [87]. Additionally, novel antioxidant compounds are emerging, such as NBP, a synthetic compound, now in a Phase III clinical trial (BAST) predicted to end in December 2022 [88]. Previously tested free radical scavengers, as described by Ebselen [89] or Edaravone [90] should be reconsidered and tested under new clinical trial guidelines for AIS (combined with reperfusion, for example).
References
- Rtf, C. A Systematic Approach to the Definition of Stroke. Austin J. Cerebrovasc. Dis. Stroke 2014, 1, 1024.
- Coupland, A.P.; Thapar, A.; Qureshi, M.I.; Jenkins, H.; Davies, A.H. The Definition of Stroke. J. R. Soc. Med. 2017, 110, 9–12.
- Sacco, R.L.; Kasner, S.E.; Broderick, J.P.; Caplan, L.R.; Connors, J.J.; Culebras, A.; Elkind, M.S.V.; George, M.G.; Hamdan, A.D.; Higashida, R.T.; et al. An Updated Definition of Stroke for the 21st Century: A Statement for Healthcare Professionals from the American Heart Association/American Stroke Association. Stroke 2013, 44, 2064–2089.
- Campbell, B.C.V.; Khatri, P. Stroke. Lancet 2020, 396, 129–142.
- Lipton, P. Ischemic Cell Death in Brain Neurons. Physiol. Rev. 1999, 79, 1431–1568.
- Murphy, S.J.; Werring, D.J. Stroke: Causes and Clinical Features. Medicine 2020, 48, 561–566.
- Katan, M.; Luft, A. Global Burden of Stroke. Semin. Neurol. 2018, 38, 208–211.
- Kuriakose, D.; Xiao, Z. Pathophysiology and Treatment of Stroke: Present Status and Future Perspectives. Int. J. Mol. Sci. 2020, 21, 7609.
- Tuo, Q.-z.; Zhang, S.-t.; Lei, P. Mechanisms of Neuronal Cell Death in Ischemic Stroke and Their Therapeutic Implications. In Medicinal Research Reviews; John Wiley and Sons Inc.: Hoboken, NJ, USA, 2021.
- Johnson, C.O.; Nguyen, M.; Roth, G.A.; Nichols, E.; Alam, T.; Abate, D.; Abd-Allah, F.; Abdelalim, A.; Abraha, H.N.; Abu-Rmeileh, N.M.; et al. Global, Regional, and National Burden of Stroke, 1990–2016: A Systematic Analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 439–458.
- Wafa, H.A.; Wolfe, C.D.A.; Emmett, E.; Roth, G.A.; Johnson, C.O.; Wang, Y. Burden of Stroke in Europe: Thirty-Year Projections of Incidence, Prevalence, Deaths, and Disability-Adjusted Life Years. Stroke 2020, 51, 2418–2427.
- Stevens, E.; Emmett, E.; Wang, Y.; McKevitt, C.; Wolfe, C.D.A. The Burden of Stroke in Europe: Report; Stroke Alliance for Europe: City Road, London, 2017.
- Kisling, L.A.; Das, J.M. Prevention Strategies; StatPearls Publ: Treasure Island, FL, USA, 2021. Available online: https://www.ncbi.nlm.nih.gov/books/NBK537222/ (accessed on 4 May 2022).
- Ntaios, G. Embolic Stroke of Undetermined Source: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2020, 75, 333–340.
- Pierik, R.; Algra, A.; van Dijk, E.; Erasmus, M.E.; van Gelder, I.C.; Koudstaal, P.J.; Luijckx, G.R.; Nederkoorn, P.J.; van Oostenbrugge, R.J.; Ruigrok, Y.M.; et al. Distribution of Cardioembolic Stroke: A Cohort Study. Cere 2020, 49, 97–104.
- Sommer, C.J. Ischemic Stroke: Experimental Models and Reality. Acta Neuropathol. 2017, 133, 245–261.
- Gormley, G.J. Chemoprevention Strategies. J. Cell. Biochem. 1992, 50, 106.
- Grysiewicz, R.A.; Thomas, K.; Pandey, D.K. Epidemiology of Ischemic and Hemorrhagic Stroke: Incidence, Prevalence, Mortality, and Risk Factors. Neurol. Clin. 2008, 26, 871–895.
- National Institute of Health. Stroke|NHLBI, NIH; National Heart, Lung and Blood institute: Bethesda, MD, USA, 2008.
- Avan, A.; Digaleh, H.; Di Napoli, M.; Stranges, S.; Behrouz, R.; Shojaeianbabaei, G.; Amiri, A.; Tabrizi, R.; Mokhber, N.; Spence, J.D.; et al. Socioeconomic Status and Stroke Incidence, Prevalence, Mortality, and Worldwide Burden: An Ecological Analysis from the Global Burden of Disease Study 2017. BMC Med. 2019, 17, 191.
- Howard, V.J.; Madsen, T.E.; Kleindorfer, D.O.; Judd, S.E.; Rhodes, J.D.; Soliman, E.Z.; Kissela, B.M.; Safford, M.M.; Moy, C.S.; McClure, L.A.; et al. Sex and Race Differences in the Association of Incident Ischemic Stroke with Risk Factors. JAMA Neurol. 2019, 76, 179–186.
- Floßmann, E.; Schulz, U.G.R.; Rothwell, P.M. Systematic Review of Methods and Results of Studies of the Genetic Epidemiology of Ischemic Stroke. Stroke 2004, 35, 212–227.
- Boehme, A.K.; Esenwa, C.; Elkind, M.S.V. Stroke Risk Factors, Genetics, and Prevention. Circ. Res. 2017, 120, 472–495.
- Deb, P.; Sharma, S.; Hassan, K.M. Pathophysiologic Mechanisms of Acute Ischemic Stroke: An Overview with Emphasis on Therapeutic Significance beyond Thrombolysis. Pathophysiology 2010, 17, 197–218.
- Hankey, G.J. Stroke. Lancet 2017, 389, 641–654.
- Barthels, D.; Das, H. Current Advances in Ischemic Stroke Research and Therapies. Biochim. Biophys. Acta—Mol. Basis Dis. 2020, 1866, 165260.
- Rosamond, W.; Flegal, K.; Furie, K.; Go, A.; Greenlund, K.; Haase, N.; Hailpern, S.M.; Ho, M.; Howard, V.; Kissela, B.; et al. Heart Disease and Stroke Statistics-2008 Update: A Report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 2008, 117, e25–e146.
- Virani, S.S.; Alonso, A.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics—2020 Update: A Report from the American Heart Association. In Circulation; Lippincott Williams & Wilkins: Hagerstown, MD, USA, 2020; pp. E139–E596.
- WHO. WHO|Stroke, Cerebrovascular Accident|Health Topics; World Health Organization: Geneva, Switzerland, 2022.
- Feske, S.K. Ischemic Stroke. Am. J. Med. 2021, 134, 1457–1464.
- Randolph, S.A. Ischemic Stroke. Work. Health Saf. 2016, 64, 444.
- Ojaghihaghighi, S.; Vahdati, S.S.; Mikaeilpour, A.; Ramouz, A. Comparison of Neurological Clinical Manifestation in Patients with Hemorrhagic and Ischemic Stroke. World J. Emerg. Med. 2017, 8, 34.
- Kuhrij, L.S.; Marang-Van De Mheen, P.J.; Van Den Berg-Vos, R.M.; De Leeuw, F.E.; Nederkoorn, P.J.; Lingsma, H.F.; De Borst, G.J.; Van Norden, A.G.W.; Eysink Smeets, M.M.; Aerden, L.A.M.; et al. Determinants of Extended Door-to-Needle Time in Acute Ischemic Stroke and Its Influence on in-Hospital Mortality: Results of a Nationwide Dutch Clinical Audit. BMC Neurol. 2019, 19, 265.
- Jamieson, D.G. Diagnosis of Ischemic Stroke. Am. J. Med. 2009, 122 (Suppl. S2), S14–S20.
- Phipps, M.S.; Cronin, C.A. Management of Acute Ischemic Stroke. BMJ 2020, 368, l6983.
- Puig, J.; Shankar, J.; Liebeskind, D.; Terceño, M.; Nael, K.; Demchuk, A.M.; Menon, B.; Dowlatshahi, D.; Leiva-Salinas, C.; Wintermark, M.; et al. From “Time Is Brain” to “Imaging Is Brain”: A Paradigm Shift in the Management of Acute Ischemic Stroke. J. Neuroimaging 2020, 30, 562–571.
- Calderon, V.J.; Kasturiarachi, B.M.; Lin, E.; Bansal, V.; Zaidat, O.O. Review of the Mobile Stroke Unit Experience Worldwide. Interv. Neurol. 2018, 7, 347–358.
- Helwig, S.A.; Ragoschke-Schumm, A.; Schwindling, L.; Kettner, M.; Roumia, S.; Kulikovski, J.; Keller, I.; Manitz, M.; Martens, D.; Grün, D.; et al. Prehospital Stroke Management Optimized by Use of Clinical Scoring vs Mobile Stroke Unit for Triage of Patients with Stroke: A Randomized Clinical Trial. JAMA Neurol. 2019, 76, 1484–1492.
- Kiewert, C.; Mdzinarishvili, A.; Hartmann, J.; Bickel, U.; Klein, J. Metabolic and Transmitter Changes in Core and Penumbra after Middle Cerebral Artery Occlusion in Mice. Brain Res. 2010, 1312, 101–107.
- Horváth, E.; Huțanu, A.; Chiriac, L.; Dobreanu, M.; Orădan, A.; Nagy, E.E. Ischemic Damage and Early Inflammatory Infiltration Are Different in the Core and Penumbra Lesions of Rat Brain after Transient Focal Cerebral Ischemia. J. Neuroimmunol. 2018, 324, 35–42.
- Shi, L.; Rocha, M.; Leak, R.K.; Zhao, J.; Bhatia, T.N.; Mu, H.; Wei, Z.; Yu, F.; Weiner, S.L.; Ma, F.; et al. A New Era for Stroke Therapy: Integrating Neurovascular Protection with Optimal Reperfusion. J. Cereb. Blood Flow Metab. 2018, 38, 2073–2091.
- Uzdensky, A.B. Apoptosis Regulation in the Penumbra after Ischemic Stroke: Expression of pro- and Antiapoptotic Proteins. In Apoptosis; Springer: Berlin/Heidelberg, Germany, 2019; pp. 687–702.
- Pawluk, H.; Woźniak, A.; Grześk, G.; Kołodziejska, R.; Kozakiewicz, M.; Kopkowska, E.; Grzechowiak, E.; Kozera, G. The Role of Selected Pro-Inflammatory Cytokines in Pathogenesis of Ischemic Stroke. In Clinical Interventions in Aging; Dove Press: Macclesfield, UK, 2020; pp. 469–484.
- Guo, Y.; Li, P.; Guo, Q.; Shang, K.; Yan, D.; Du, S.; Lu, Y. Pathophysiology and Biomarkers in Acute Ischemic Stroke—A Review. Trop. J. Pharm. Res. 2013, 12, 1097–1105.
- Mergenthaler, P.; Dirnagl, U.; Kunz, A. Ischemic Stroke: Basic Pathophysiology and Clinical Implication. In Neuroscience in the 21st Century—From Basic to Clinical; Springer: New York, NY, USA, 2013; pp. 2543–2563, Chapter 97.
- Singhal, A.B.; Lo, E.H.; Dalkara, T.; Moskowitz, M.A. Ischemic Stroke: Basic Pathophysiology and Neuroprotective Strategies. Acute Ischemic Stroke—Imaging and Intervention; Springer: Berlin/Heidelberg, Germany, 2011; pp. 1–24, Chapter 1.
- Nour, M.; Scalzo, F.; Liebeskind, D.S. Ischemia-Reperfusion Injury in Stroke. Interv. Neurol. 2013, 1, 185–199.
- Campbell, B.C.V.; Meretoja, A.; Donnan, G.A.; Davis, S.M. Twenty-Year History of the Evolution of Stroke Thrombolysis with Intravenous Alteplase to Reduce Long-Term Disability. Stroke 2015, 46, 2341–2346.
- Molina, C.A.; Saver, J.L. Extending Reperfusion Therapy for Acute Ischemic Stroke: Emerging Pharmacological, Mechanical, and Imaging Strategies. Stroke 2005, 36, 2311–2320.
- Bhaskar, S.; Stanwell, P.; Cordato, D.; Attia, J.; Levi, C. Reperfusion Therapy in Acute Ischemic Stroke: Dawn of a New Era? BMC Neurol. 2018, 18, 1–26.
- Daniel, M.; Goel, I.; Sakai, Y.; Teramura, Y. Current Status of Ischemic Stroke Treatment: From Thrombolysis to Potential Regenerative Medicine. Regen. Ther. 2021, 18, 408–417.
- Herpich, F.; Rincon, F. Management of Acute Ischemic Stroke. Crit. Care Med. 2020, 48, 1654–1663.
- Campbell, B.C.V.; Mitchell, P.J.; Churilov, L.; Yassi, N.; Kleinig, T.J.; Dowling, R.J.; Yan, B.; Bush, S.J.; Dewey, H.M.; Thijs, V.; et al. Tenecteplase versus Alteplase before Thrombectomy for Ischemic Stroke. N. Engl. J. Med. 2018, 378, 1573–1582.
- Berge, E.; Whiteley, W.; Audebert, H.; De Marchis, G.M.; Fonseca, A.C.; Padiglioni, C.; de la Ossa, N.P.; Strbian, D.; Tsivgoulis, G.; Turc, G. European Stroke Organisation (ESO) Guidelines on Intravenous Thrombolysis for Acute Ischaemic Stroke. Eur. Stroke J. 2021, 6, I.
- Seners, P.; Caroff, J.; Chausson, N.; Turc, G.; Denier, C.; Piotin, M.; Aghasaryan, M.; Alecu, C.; Chassin, O.; Lapergue, B.; et al. Recanalization before Thrombectomy in Tenecteplase vs. Alteplase-Treated Drip-and-Ship Patients. J. Stroke 2019, 21, 105–107.
- Papanagiotou, P.; Ntaios, G. Endovascular Thrombectomy in Acute Ischemic Stroke. Circ. Cardiovasc. Interv. 2018, 11, e005362.
- Blanc, R.; Escalard, S.; Baharvadhat, H.; Desilles, J.P.; Boisseau, W.; Fahed, R.; Redjem, H.; Ciccio, G.; Smajda, S.; Maier, B.; et al. Recent Advances in Devices for Mechanical Thrombectomy. Expert Rev. Med. Devices 2020, 17, 697–706.
- Maurice, A.; Ferré, J.C.; Ronzière, T.; Devys, J.M.; Subileau, A.; Laffon, M.; Laviolle, B.; Beloeil, H. GASS Trial Study Protocol: A Multicentre, Single-Blind, Randomised Clinical Trial Comparing General Anaesthesia and Sedation during Intra-Arterial Treatment for Stroke. BMJ Open 2019, 9, e024249.
- Tsivgoulis, G.; Katsanos, A.H. Important Advances in Stroke Research in 2020. Lancet Neurol. 2021, 20, 2–3.
- Yang, P.; Zhang, Y.; Zhang, L.; Zhang, Y.; Treurniet, K.M.; Chen, W.; Peng, Y.; Han, H.; Wang, J.; Wang, S.; et al. Endovascular Thrombectomy with or without Intravenous Alteplase in Acute Stroke. N. Engl. J. Med. 2020, 382, 1981–1993.
- Katsanos, A.H.; Malhotra, K.; Goyal, N.; Arthur, A.; Schellinger, P.D.; Köhrmann, M.; Krogias, C.; Turc, G.; Magoufis, G.; Leys, D.; et al. Intravenous Thrombolysis Prior to Mechanical Thrombectomy in Large Vessel Occlusions. Ann. Neurol. 2019, 86, 395–406.
- Nogueira, R.G.; Tsivgoulis, G. Large Vessel Occlusion Strokes after the DIRECT-MT and SKIP Trials: Is the Alteplase Syringe Half Empty or Half Full? Stroke 2020, 51, 3182–3186.
- Jeyaseelan, K.; Lim, K.Y.; Armugam, A. Neuroprotectants in Stroke. Pharmacotherapy 2008, 9, 887–900.
- DeGraba, T.J.; Pettigrew, L.C. Why Do Neuroprotective Drugs Work in Animals but Not Humans? Neurol. Clin. 2000, 18, 475–493.
- Dirnagl, U.; Priller, J. Focal Cerebral Ischemia: The Multifaceted Role of Glial Cells. In Neuroglia; Oxford Academic: Oxford, UK, 2013.
- Nedergaard, M.; Dirnagl, U. Role of Glial Cells in Cerebral Ischemia. Glia 2005, 50, 281–286.
- Wang, L.; Xiong, X.; Zhang, L.; Shen, J. Neurovascular Unit: A Critical Role in Ischemic Stroke. CNS Neurosci. Ther. 2021, 27, 7–16.
- Luo, Y.; Tang, H.; Li, H.; Zhao, R.; Huang, Q.; Liu, J. Recent Advances in the Development of Neuroprotective Agents and Therapeutic Targets in the Treatment of Cerebral Ischemia. Eur. J. Med. Chem. 2019, 162, 132–146.
- Lapchak, P.A.; Zhang, J.H. The High Cost of Stroke and Stroke Cytoprotection Research. Transl. Stroke Res. 2016, 8, 307–317.
- Fisher, M.; Savitz, S.I. Pharmacological Brain Cytoprotection in Acute Ischaemic Stroke—Renewed Hope in the Reperfusion Era. Nat. Rev. Neurol. 2022, 18, 193–202.
- Cheng, Y.D.; Al-Khoury, L.; Zivin, J.A. Neuroprotection for Ischemic Stroke: Two Decades of Success and Failure. NeuroRx 2004, 1, 36.
- Das, J.M.; Zito, P.M. Nimodipine. In xPharm: The Comprehensive Pharmacology Reference; Elsevier Inc.: Amsterdam, The Netherlands, 2021; pp. 1–7.
- Horn, J.; De Haan, R.J.; Vermeulen, M.; Limburg, M. Very Early Nimodipine Use in Stroke (VENUS). Stroke 2001, 32, 461–465.
- Magnesium and Verapamil after Recanalization in Ischemia of the Cerebrum: A Clinical and Translational Study (MAVARIC; NCT02912663). Available online: https://clinicaltrials.gov/ct2/results?cond=&term=NCT02912663&cntry=&state=&city=&dist= (accessed on 7 June 2022).
- Saver, J.L.; Starkman, S.; Eckstein, M.; Stratton, S.J.; Pratt, F.D.; Hamilton, S.; Conwit, R.; Liebeskind, D.S.; Sung, G.; Kramer, I.; et al. Prehospital Use of Magnesium Sulfate as Neuroprotection in Acute Stroke. N. Engl. J. Med. 2015, 372, 528–536.
- Cigel, A.; Sayin, O.; Gurgen, S.G.; Sonmez, A. Long Term Neuroprotective Effects of Acute Single Dose MK-801treatment against Traumatic Brain Injury in Immature Rats. Neuropeptides 2021, 88, 102161.
- Sattler, R.; Xiong, Z.; Lu, W.Y.; Hafner, M.; MacDonald, J.F.; Tymianski, M. Specific Coupling of NMDA Receptor Activation to Nitric Oxide Neurotoxicity by PSD-95 Protein. Science 1999, 284, 1845–1848.
- Ugalde-Triviño, L.; Díaz-Guerra, M. PSD-95: An Effective Target for Stroke Therapy Using Neuroprotective Peptides. Int. J. Mol. Sci. 2021, 22, 12585.
- Aarts, M.; Liu, Y.; Liu, L.; Besshoh, S.; Arundine, M.; Gurd, J.W.; Wang, Y.T.; Salter, M.W.; Tymianski, M. Treatment of Ischemic Brain Damage by Perturbing NMDA Receptor-PSD-95 Protein Interactions. Science 2002, 298, 846–850.
- Hill, M.D.; Goyal, M.; Menon, B.K.; Nogueira, R.G.; McTaggart, R.A.; Demchuk, A.M.; Poppe, A.Y.; Buck, B.H.; Field, T.S.; Dowlatshahi, D.; et al. Efficacy and Safety of Nerinetide for the Treatment of Acute Ischaemic Stroke (ESCAPE-NA1): A Multicentre, Double-Blind, Randomised Controlled Trial. Lancet 2020, 395, 878–887.
- Fisher, M.; Israel, B.; Xiong, Y.; Wakhloo, A.K. Advances in Acute Ischemic Stroke Therapy. Circ. Res. 2022, 130, 1230–1251.
- Efficacy and Safety of Nerinetide in Participants with Acute Ischemic Stroke Undergoing Endovascular Thrombectomy Excluding Thrombolysis.(ESCAPE-NEXT; NCT04462536). Available online: https://clinicaltrials.gov/ct2/results?cond=&term=NCT04462536&cntry=&state=&city=&dist= (accessed on 7 June 2022).
- Bennion, D.M.; Isenberg, J.D.; Harmel, A.T.; DeMars, K.; Dang, A.N.; Jones, C.H.; Pignataro, M.E.; Graham, J.T.; Steckelings, U.M.; Alexander, J.C.; et al. Post-Stroke Angiotensin II Type 2 Receptor Activation Provides Long-Term Neuroprotection in Aged Rats. PLoS ONE 2017, 12, e0180738.
- Eklund, P.C.; Långvik, O.K.; Wärnå, J.P.; Salmi, T.O.; Willför, S.M.; Sjöholm, R.E. Chemical Studies on Antioxidant Mechanisms and Free Radical Scavenging Properties of Lignans. Org. Biomol. Chem. 2005, 3, 3336–3347.
- Aliena-Valero, A.; Rius-Pérez, S.; Baixauli-Martín, J.; Torregrosa, G.; Chamorro, Á.; Pérez, S.; Salom, J.B. Uric Acid Neuroprotection Associated to IL-6/STAT3 Signaling Pathway Activation in Rat Ischemic Stroke. Mol. Neurobiol. 2020, 58, 408–423.
- Chamorro, Á.; Amaro, S.; Castellanos, M.; Segura, T.; Arenillas, J.; Martí-Fábregas, J.; Gállego, J.; Krupinski, J.; Gomis, M.; Cánovas, D.; et al. Safety and Efficacy of Uric Acid in Patients with Acute Stroke (URICO-ICTUS): A Randomised, Double-Blind Phase 2b/3 Trial. Lancet Neurol. 2014, 13, 453–460.
- Chamorro, Á.; Lo, E.H.; Renú, A.; Van Leyden, K.; Lyden, P.D. The Future of Neuroprotection in Stroke. J. Neurol. Neurosurg. Psychiatry 2021, 92, 129–135.
- Efficacy and Safety of Butylphthalide for Acute Ischemic Stroke Patients Receiving Intravenous Thrombolysis or Endovascular Treatment. (BAST; NCT03539445). Available online: https://clinicaltrials.gov/ct2/results?cond=&term=NCT03539445&cntry=&state=&city=&dist= (accessed on 7 June 2022).
- Yamaguchi, T.; Sano, K.; Takakura, K.; Saito, I.; Shinohara, Y.; Asano, T.; Yasuhara, H. Ebselen in Acute Ischemic Stroke. Stroke 1998, 29, 12–17.
- Otomo, E.; Tohgi, H.; Kogure, K.; Hirai, S.; Takakura, K.; Terashi, A.; Gotoh, F.; Maruyama, S.; Tazaki, Y.; Shinohara, Y.; et al. Effect of a Novel Free Radical Scavenger, Edaravone (MCI-186), on Acute Brain Infarction. Randomized, Placebo-Controlled, Double-Blind Study at Multicenters. Cerebrovasc. Dis. 2003, 15, 222–229.
More
Information
Subjects:
Neurosciences
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
3.9K
Revisions:
2 times
(View History)
Update Date:
20 Oct 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No