 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Lorry Carrié | + 2097 word(s) | 2097 | 2020-11-05 09:36:22 |
Video Upload Options
Melanoma is a devastating skin cancer characterized by an impressive metabolic plasticity. Melanoma cells are able to adapt to the tumor microenvironment by using a variety of fuels that contribute to tumor growth and progression. In this review, the authors summarize the contribution of the lipid metabolic network in melanoma plasticity and aggressiveness, with a particular attention to specific lipid classes such as glycerophospholipids, sphingolipids, sterols and eicosanoids. They also highlight the role of adipose tissue in tumor progression as well as the potential antitumor role of drugs targeting critical steps of lipid metabolic pathways in the context of melanoma.
1. Introduction
The metabolic remodeling is a crucial process that allows melanoma cells to adapt to tumor microenvironment (TME) and to sustain growth and dissemination [1][2]. A comparative metabolic flux profiling of melanoma cell lines and normal melanocytes showed that all melanoma cells consumed more glucose and produced more lactate than melanocytes [3]. Interestingly, emerging evidence reported numerous alterations of the lipid metabolic network that could sustain cell growth and metastasis in melanoma cells (Figure 1).
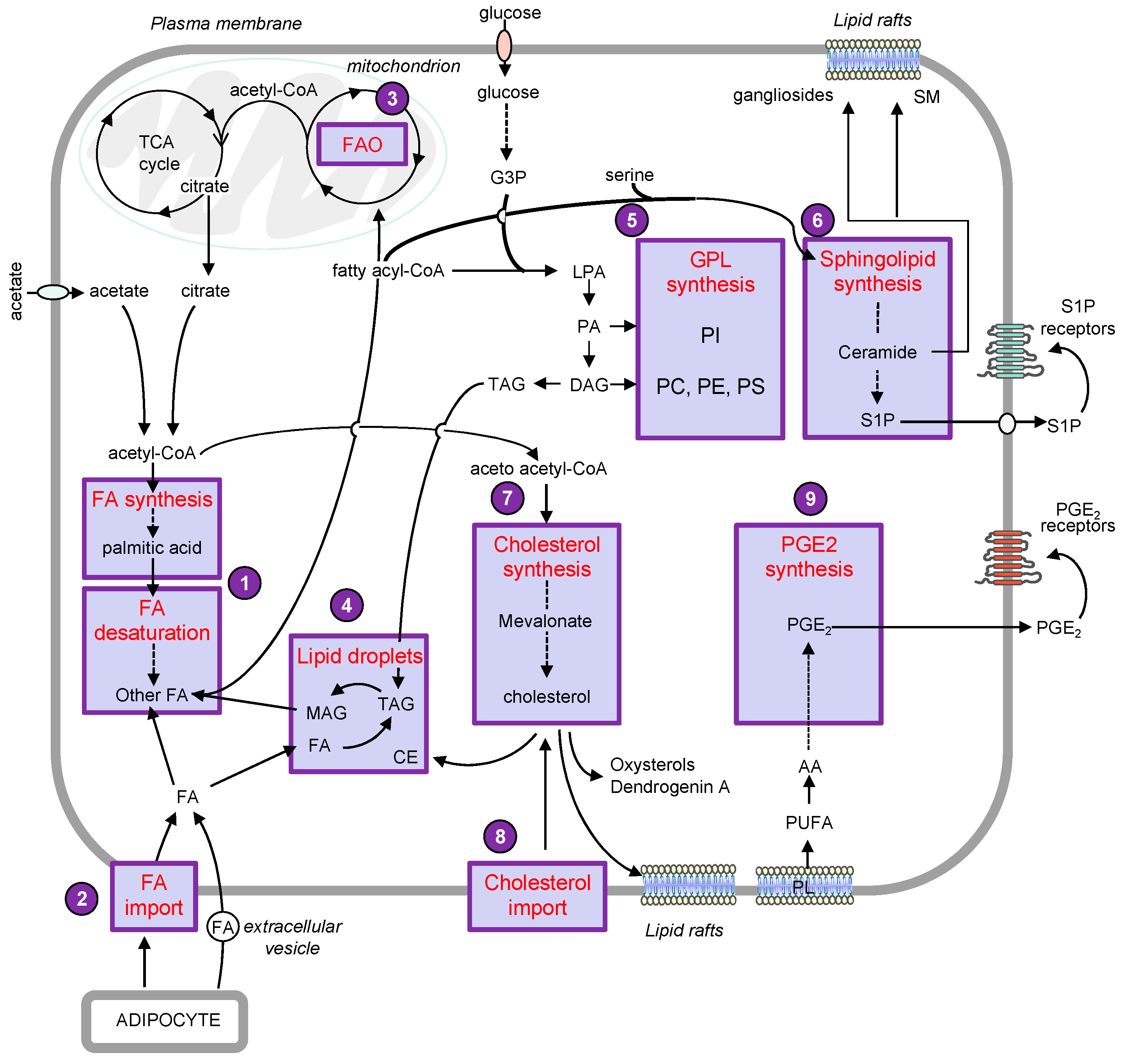
2. Lipid metabolism and melanoma progression
The most prominent phenomenon is an increased rate of lipogenesis, in which nutrient-derived carbons get converted into fatty acids (FAs), sterols and complex lipids. Lipogenesis relies mainly on the availability of acetyl-CoA. The main precursor of cytosolic acetyl-CoA is citrate originating from the tricarboxylic acid (TCA) cycle under normal conditions [4]. This conversion is catalyzed by ATP citrate lyase (ACLY), which is overexpressed in a variety of cancer types, including melanoma. Moreover, increased ACLY expression was associated with poor outcome of patients with melanoma [5][6]. During metabolic stress such as hypoxia, the synthesis of acetyl-CoA preferentially originates from acetate [7][8]. Acetate dependence is specific to BRAF mutant but not NRAS mutant or wild-type BRAF/NRAS melanoma cells [9]. Importantly, melanoma brain metastases, which are associated with an extremely poor prognosis, have been shown to exhibit increased dependency on acetate [10].
FA synthesis starts with the carboxylation of acetyl-CoA to malonyl-CoA, which is catalyzed by acetyl-CoA carboxylase (ACC1). Then, through a series of repetitive condensations catalyzed by the FA synthase (FASN), molecules of malonyl-CoA are assembled to form palmitic acid. The expression of ACC1 [11] and FASN [12] is upregulated in human melanoma, as compared to conventional nevi. The increased expression of FASN occurs independently of the BRAF and NRAS mutation status [5] but is associated with the Breslow thickness and poor prognosis [12][13]. The specific inhibition of FASN activity with the anti-obesity drug Orlistat was reported to reduce the occurrence and number of lung metastases in a murine model of melanoma [14]. Thereafter, elongation and desaturation of palmitic acid generate the basis for a diverse spectrum of saturated and unsaturated FA that can be activated into fatty acyl-CoA by acyl-CoA synthetase long-chain (ACSL) family members. Of note, the expression of ACSL3 has been also associated to a worse prognosis in melanoma [15]. Moreover, a recent study reported that oleic acid, an abundant FA in lymph, protected melanoma cells from ferroptosis in an ACSL3-dependent manner and increased their capacity to form metastasis [16]. Once activated, the FA can be incorporated into triglycerides (also named triacylglycerols (TAGs)), glycerophospholipids (GPL) and sphingolipids (SL) or undergo β-oxidation in mitochondria for energy generation [17]. In addition to their role in fueling various lipid metabolisms, FAs also participate to protein acylation, thereby controlling protein trafficking, membrane localization and signaling activities [18]. For instance, the S-palmitoylation of the melanocortin-1 receptor (MC1R), which corresponds to the covalent attachment of palmitic acid to the protein at cysteine residues, was associated with MC1R activation, thereby reducing melanomagenesis in mice [19]. Conversely, the S-palmitoylation of the TEA domain (TEAD) transcription factors was shown to be critical in TEAD’s binding to the Hippo kinases YAP (Yes-associated protein) and TAZ (Transcriptional activator with PDZ domain) [20]. The YAP/TAZ-TEAD complex is known to activate expression of several genes that favor tumor growth and metastasis in various solid cancers, including melanoma [21].
Beside FA synthesis, the cytosolic acetyl-CoA can also be transformed into 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA), which is then converted into mevalonate by the HMG-CoA reductase (HMGCR), the rate-limiting step of cholesterol biosynthesis. Analysis of public databases revealed that ~60% of melanomas had increased expression (including chromosomal copy number increases) in at least one of the cholesterol synthesis genes. These events were associated with decreased melanoma patient survival [22].
While de novo lipogenesis constitutes a valuable source of energy, as well as lipid mediators, hypoxia or driver mutations can also prime melanoma cells to consume FA from the TME, via FA β-oxidation (FAO), to meet their energetic demands [23]. FAO was reported to promote melanoma progression. For instance, carnitine palmitoyltransferase 2 (CPT2), which is critical for translocation of long-chain acyl-CoA into the mitochondrial matrix, is one of the most significantly upregulated genes in melanoma as compared to benign nevi [24]. Moreover, thanks to a targeted analysis of human tumor samples from the TCGA database, it was recently revealed that increased expression of FAO enzymes correlated with poor overall survival in melanoma patients [25]. In accordance, it was demonstrated that FAO contributed significantly to the energy reserves of metastatic 4C11+ cells, which were derived from melan-a melanocytes after sequential detachment-re-adhesion cycles [26]. How FAO promotes melanoma progression is still unclear. One can imagine that FAs serve as a valuable source of acetyl-CoA that contributes to citrate formation, after entering the TCA cycle, and provide an ATP boost for tumor cells under nutrient-depleted conditions [27]. Interestingly, other studies in which melanoma cells were co-cultured with adipocytes have shown that adipocyte-derived lipids were utilized in the FAO pathway and decreased the dependence on de novo lipogenesis [25][28]. In this context, glucose oxidation and lactate release were unchanged, indicating that glycolysis was not impacted [29].
FA can be imported from plasma and lymph [16] either through the FA translocase (FAT/CD36), the plasma membrane-associated FA binding proteins (FABP) or the FA transport proteins (FATP), and FA levels can increase through close contact with adipocytes [28][30]. This latter event was inhibited by the FATP inhibitor lipofermata in a zebrafish melanoma model [28]. Importantly, a recent study reported that, when melanoma cells were exposed to the aged fibroblast lipid secretome, they increased FA uptake via FATP2, whose expression was upregulated. Inhibiting FATP2 with lipofermata was shown to overcome age-related resistance to BRAF/MEK inhibition in animal models and significantly extend survival in older animals [31]. Moreover, FABP7 has been associated with increased proliferation and invasive properties of melanoma cells [32][33][34]. CD36-mediated FA uptake is prominent in metastasis-initiating melanoma cells, and this change was correlated with poor prognosis in melanoma patients [35], thereby highlighting the importance of FA uptake for melanoma progression.
Increased FA biosynthesis and FA uptake may lead to increased levels of multiple lipids with a signaling function that can affect numerous cellular processes, including melanoma cell differentiation and motility. Melanoma is notorious for its high metastatic potential. Melanoma invasive behavior is controlled by signaling pathways, e.g., the canonical and non-canonical Wingless-type (Wnt) and the transforming growth factor beta (TGF-β) signaling pathways, that have been described to not only regulate the actin cytoskeleton but also the expression levels and the function of the lineage commitment factor microphthalmia-associated transcription factor (MITF) [36]. A wide range of cellular stresses including hypoxia [37][38][39], low glucose [40] and inflammatory signaling [41][42][43] were shown to reduce MITF expression and increase the metastatic properties of melanoma cells. Moreover, meta-analysis of gene expression profiling of hundreds of human melanoma cells identified a highly invasive phenotype, characterized by extremely low MITF expression, associated with a stemness- and epithelial-to-mesenchymal transition (EMT)-based gene expression signature [44][45][46]. It is now well recognized that melanoma cells are highly plastic and can undergo phenotype switching that contributes to tumor progression. During this process, melanoma cells with an MITF-low phenotype undergo invasion and dissemination, and then switch back to an MITF-high phenotype at the metastatic site in order to proliferate [47]. Importantly, the reduction of MITF expression has been associated with a switch in EMT-associated transcription factors (EMT-TFs). In particular, a reduced expression of ZEB2 and SNAIL2, in favor of an increased expression in ZEB1 and TWIST1, has been linked to MITF downregulation, E-cadherin loss and increased invasive properties of human melanoma cells [48].
Interestingly, recent findings revealed that the lipogenic enzyme ACLY regulated MITF, and its downstream transcriptional targets by controlling histone acetylation at its promoter [6]. Moreover, low activity of stearoyl-CoA desaturase (SCD), which catalyzes the rate-limiting step of FA desaturation, reduced MITF expression and maintained melanoma cells in an MITF-low de-differentiated state [49]. Inversely, MITF was identified as a regulator of SCD expression and FA saturation, thereby establishing a positive feedback loop to stabilize an MITF-low state associated with increased metastatic dissemination. Mechanistically, low SCD expression and activity promoted ER stress and the phosphorylation of eukaryotic initiation factor-α (eIF2α) leading to the activation of an ATF4- and NF-κB-dependent inflammatory signaling that sustains a reduced MITF expression and melanoma cell dedifferentiation [49]. These data demonstrate that FA metabolism can regulate melanoma cell differentiation and progression.
This review aims to illustrate the major alterations affecting lipid storage organelles and the metabolism of the main lipid classes during melanoma development (see Figure 2 for a detailed view) and how these metabolic dysregulations contribute to phenotype plasticity and/or melanoma aggressiveness. How these metabolic vulnerabilities could be targeted for therapeutic benefit is also highlighted.
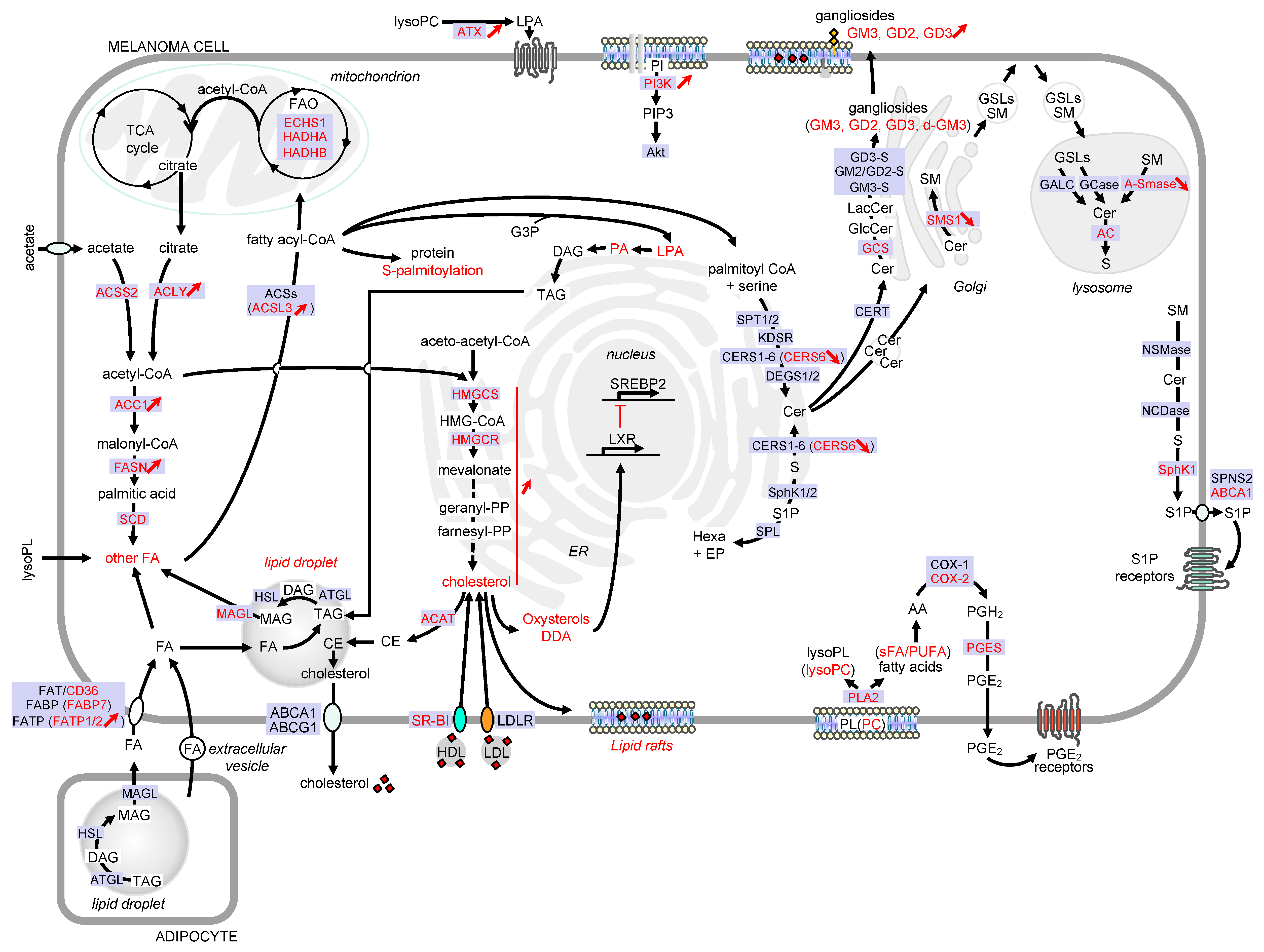
Figure 2. Detailed view of the major alterations of lipid storage and metabolism pathways during melanoma development. Only lipid pathways with reported modifications in melanoma are represented. Enzymes, receptors and transporters are indicated in blue boxes. Modifications in melanoma are highlighted in red. Abbreviations: AA, arachidonic acid; ABC, ATP-binding cassette transporter; AC, acid ceramidase; ACAT, acyl-CoA: cholesterol acyltransferase; ACC, acetyl-CoA carboxylase; ACLY, ATP citrate lyase; ACS, acyl-CoA synthetase; ACSL3, acyl-CoA synthetase long chain 3; Akt, AKT serine/threonine kinase; A-SMase, acid sphingomyelinase; ATX, lysophospholipase D autotaxin; CE, cholesteryl ester; Cer, ceramide; CERS, ceramide synthase; CERT, ceramide transport protein; COX, cyclooxygenase; DAG, diacylglycerol; DDA, dendrogenin A; DEGS, dihydroceramide desaturase; d-GM3, de-N-acetyl GM3; ECHS1, enoyl-CoA hydratase short chain 1; EP, ethanolamine 1-phosphate; ER, endoplasmic reticulum; FA, fatty acid; FAO, fatty acid -oxidation; FASN, fatty acid synthase; FAT, fatty acid translocase; FABP, fatty acid binding protein; FATP, fatty acid transport protein; GALC, galactosylceramidase; GCase, glucosylceramidase; GCS, glucosylceramide synthase; GD3-S, GD3 synthase; GM2/GD2-S, GD2/GM2 synthase; GlcCer, glucosylceramide; GM3-S, GM3 synthase; GSL, glycosphingolipid; G3P, glycerol-3-phosphate; HADHA, hydroxyacyl-CoA dehydrogenase subunit alpha; HADHB, hydroxyacyl-CoA dehydrogenase subunit beta; hexa, hexadecenal; HMG-CoA, 3-hydroxy-3-methylglutaryl-CoA; HMGCR, HMG-CoA reductase; HMGCS, HMG-CoA synthase; KDSR, 3-ketosphinganine reductase; LacCer, lactosylceramide; LDL, low-density lipoprotein; LDLR, low-density lipoprotein receptor; LPA, lysophosphatidic acid; lysoPC, lysophosphatidylcholine; lysoPL, lysophospholipid; LXR, liver X receptors; MAG, monoacylglycerol; MAGL, monoacylglycerol lipase; NCDase, neutral ceramidase; NSMase, neutral sphingomyelinase; PA, phosphatidic acid; PC, phosphatidylcholine; PGE2, prostaglandin E2; PGES, prostaglandin E synthase; PGH2, prostaglandin H2; PI, phosphatidylinositol; PI3K, phosphatidylinositol-3-kinase; PIP3, phosphatidylinositol-3,4,5-triphosphate; PL, phospholipid; PLA2, phospholipase A2; PUFA, polyunsaturated fatty acid; S, sphingosine; SCD, stearoyl-CoA desaturase; sFA, saturated fatty acid; SM, sphingomyelin; SMS, sphingomyelin synthase; SphK, sphingosine kinase; SPL, sphingosine 1-phosphate lyase; SPNS2, sphingolipid transporter 2; SPT, serine palmitoyltransferase; SR-BI, scavenger receptor class B type I; SREBP2, sterol regulatory element binding protein 2; S1P, sphingosine 1-phosphate; TAG, triacylglycerol; TCA, tricarboxylic acid.
References
- Franziska Baenke; Nathalie Dhomen; Eyal Gottlieb; Richard Marais; Melanoma Metabolism. Melanoma 2019, -, 1-24, 10.1007/978-1-4614-7322-0_28-2.
- Angelica Avagliano; Giuseppe Fiume; Alessandra Pelagalli; Gennaro Sanità; Maria Rosaria Ruocco; Stefania Montagnani; Alessandro Arcucci; Metabolic Plasticity of Melanoma Cells and Their Crosstalk With Tumor Microenvironment. Frontiers in Oncology 2020, 10, 722, 10.3389/fonc.2020.00722.
- David A. Scott; Adam D. Richardson; Fabian V. Filipp; Christine A. Knutzen; Gary G. Chiang; Ze'ev A. Ronai; Andrei L. Osterman; Jeffrey W. Smith; Comparative Metabolic Flux Profiling of Melanoma Cell Lines. Journal of Biological Chemistry 2011, 286, 42626-42634, 10.1074/jbc.m111.282046.
- Grant M. Fischer; Y. N. Vashisht Gopal; Jennifer L. McQuade; Weiyi Peng; Ralph J. DeBerardinis; Michael A. Davies; Metabolic strategies of melanoma cells: Mechanisms, interactions with the tumor microenvironment, and therapeutic implications. Pigment Cell & Melanoma Research 2017, 31, 11-30, 10.1111/pcmr.12661.
- Su Wu; Anders M. Näär; SREBP1-dependent de novo fatty acid synthesis gene expression is elevated in malignant melanoma and represents a cellular survival trait. Scientific Reports 2019, 9, 1-17, 10.1038/s41598-019-46594-x.
- Weinan Guo; Jinyuan Ma; Yuqi Yang; Sen Guo; Weigang Zhang; Tao Zhao; Xiuli Yi; Huina Wang; Shiyu Wang; Yu Liu; et al.Wei DaiXuguang ChenQiong ShiGang WangTianwen GaoChunying Li ATP-Citrate Lyase Epigenetically Potentiates Oxidative Phosphorylation to Promote Melanoma Growth and Adaptive Resistance to MAPK Inhibition. Clinical Cancer Research 2020, 26, 2725-2739, 10.1158/1078-0432.ccr-19-1359.
- Jurre J. Kamphorst; Michelle K Chung; Jing Fan; Joshua D. Rabinowitz; Quantitative analysis of acetyl-CoA production in hypoxic cancer cells reveals substantial contribution from acetate. Cancer & Metabolism 2014, 2, 23, 10.1186/2049-3002-2-23.
- Yukie Yoshii; Takako Furukawa; Tsuneo Saga; Yasuhisa Fujibayashi; Acetate/acetyl-CoA metabolism associated with cancer fatty acid synthesis: Overview and application. Cancer Letters 2015, 356, 211-216, 10.1016/j.canlet.2014.02.019.
- Alexander J. Lakhter; James Hamilton; Raymond L. Konger; Nickolay Brustovetsky; Hal E. Broxmeyer; Samisubbu R. Naidu; Glucose-independent Acetate Metabolism Promotes Melanoma Cell Survival and Tumor Growth. Journal of Biological Chemistry 2016, 291, 21869-21879, 10.1074/jbc.m115.712166.
- Tomoyuki Mashimo; Kumar Pichumani; Vamsidhara Vemireddy; Kimmo J. Hatanpaa; Dinesh Kumar Singh; Shyam Sirasanagandla; Suraj Nannepaga; Sara G. Piccirillo; Zoltan Kovacs; Chan Foong; et al.Zhiguang HuangSamuel BarnettBruce E. MickeyRalph J. DeBerardinisBenjamin P. TuElizabeth A. MaherRobert M. Bachoo Acetate Is a Bioenergetic Substrate for Human Glioblastoma and Brain Metastases. Cell 2014, 159, 1603-1614, 10.1016/j.cell.2014.11.025.
- Wenjuan Li; Chunjing Zhang; Hongyan Du; Vincent Huang; Brandi Sun; John P. Harris; Quitin Richardson; Xinggui Shen; Rong Jin; Guohong Li; et al.Christopher G. KevilXin GuRunhua ShiYunfeng Zhao Withaferin A suppresses the up-regulation of acetyl-coA carboxylase 1 and skin tumor formation in a skin carcinogenesis mouse model. Molecular Carcinogenesis 2015, 55, 1739-1746, 10.1002/mc.22423.
- Payal Kapur; Dinesh Rakheja; Lonnie C Roy; Mai P Hoang; Fatty acid synthase expression in cutaneous melanocytic neoplasms. Modern Pathology 2005, 18, 1107-1112, 10.1038/modpathol.3800395.
- Daniele Innocenzi; P. L. Alò; A. Balzani; V. Sebastiani; V. Silipo; Giuseppe La Torre; G. Ricciardi; C. Bosman; Stefano Calvieri; Fatty acid synthase expression in melanoma. Journal of Cutaneous Pathology 2003, 30, 23-28, 10.1034/j.1600-0560.2003.300104.x.
- Marco A. Carvalho; Karina G. Zecchin; Fabiana Seguin; Débora C. Bastos; Michelle Agostini; Ana Lúcia C.A. Rangel; Sílvio S. Veiga; Helena F. Raposo; Helena C.F. Oliveira; Massimo Loda; et al.Ricardo D. ColettaEdgard Graner Fatty acid synthase inhibition with Orlistat promotes apoptosis and reduces cell growth and lymph node metastasis in a mouse melanoma model. International Journal of Cancer 2008, 123, 2557-2565, 10.1002/ijc.23835.
- Wei-Ching Chen; Chih-Yang Wang; Yu-Hsuan Hung; Tzu-Yang Weng; Meng-Chi Yen; Ming-Derg Lai; Systematic Analysis of Gene Expression Alterations and Clinical Outcomes for Long-Chain Acyl-Coenzyme A Synthetase Family in Cancer. PLoS ONE 2016, 11, e0155660, 10.1371/journal.pone.0155660.
- Jessalyn M. Ubellacker; Alpaslan Tasdogan; Vijayashree Ramesh; Bo Shen; Evann C. Mitchell; Misty S. Martin-Sandoval; Zhimin Gu; Michael L. McCormick; Alison B. Durham; Douglas R. Spitz; et al.Zhiyu ZhaoThomas P. MathewsSean J. Morrison Lymph protects metastasizing melanoma cells from ferroptosis. Nature 2020, 585, 113-118, 10.1038/s41586-020-2623-z.
- Claudio R. Santos; Almut Schulze; Lipid metabolism in cancer. FEBS Journal 2012, 279, 2610-2623, 10.1111/j.1742-4658.2012.08644.x.
- Baoen Chen; Yang Sun; Jixiao Niu; Gopala K. Jarugumilli; Xu Wu; Protein Lipidation in Cell Signaling and Diseases: Function, Regulation, and Therapeutic Opportunities. Cell Chemical Biology 2018, 25, 817-831, 10.1016/j.chembiol.2018.05.003.
- Shuyang Chen; Bo Zhu; Chengqian Yin; Wei Liu; Changpeng Han; Baoen Chen; Tongzheng Liu; Xin Li; Xiang Chen; Chunying Li; et al.Chen ShuyangJun ZhouZhi-Xiang XuXiumei GaoXu WuColin R. GodingRutao Cui Palmitoylation-dependent activation of MC1R prevents melanomagenesis. Nature 2017, 549, 399-403, 10.1038/nature23887.
- PuiYee Chan; Xiao Han; Baohui Zheng; Michael DeRan; Jianzhong Yu; Gopala K. Jarugumilli; Hua Deng; Duojia Pan; Xuelian Luo; Xu Wu; et al. Autopalmitoylation of TEAD proteins regulates transcriptional output of the Hippo pathway. Nature Chemical Biology 2016, 12, 282-289, 10.1038/nchembio.2036.
- Barry J. Thompson; YAP/TAZ: Drivers of Tumor Growth, Metastasis, and Resistance to Therapy. BioEssays 2020, 42, e1900162, 10.1002/bies.201900162.
- Omer F. Kuzu; Mohammad A. Noory; Gavin P. Robertson; The Role of Cholesterol in Cancer. Cancer Research 2016, 76, 2063-2070, 10.1158/0008-5472.can-15-2613.
- Jurre J. Kamphorst; Justin R. Cross; Jing Fan; Elisa De Stanchina; Robin Mathew; Eileen P. White; Craig B. Thompson; Joshua D. Rabinowitz; Hypoxic and Ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proceedings of the National Academy of Sciences 2013, 110, 8882-8887, 10.1073/pnas.1307237110.
- Venil N. Sumantran; Pratik Mishra; N Sudhakar; Microarray analysis of differentially expressed genes regulating lipid metabolism during melanoma progression.. Indian J. Biochem. Biophys. 2015, 52, 125-31.
- Emily Clement; Ikrame Lazar; Camille Attané; Lorry Carrié; Stéphanie Dauvillier; Manuelle Ducoux‐Petit; David Esteve; Thomas Menneteau; Mohamed Moutahir; Sophie Le Gonidec; et al.Stéphane DallePhilippe ValetOdile Burlet‐SchiltzCatherine MullerLaurence Nieto Adipocyte extracellular vesicles carry enzymes and fatty acids that stimulate mitochondrial metabolism and remodeling in tumor cells. The EMBO Journal 2020, 39, e102525, 10.15252/embj.2019102525.
- Mariana Figueiredo Rodrigues; Emilie Obre; Fabiana H.M. De Melo; Jr Gilson C. Santos; Antonio Galina; Miriam Galvonas Jasiulionis; Rodrigue Rossignol; Franklin David Rumjanek; Nivea Dias Amoedo; Enhanced OXPHOS, glutaminolysis and β-oxidation constitute the metastatic phenotype of melanoma cells. Biochemical Journal 2016, 473, 703-715, 10.1042/bj20150645.
- Arkaitz Carracedo; Lewis C. Cantley; Pier Paolo Pandolfi; Cancer metabolism: fatty acid oxidation in the limelight. Nature Reviews Cancer 2013, 13, 227-232, 10.1038/nrc3483.
- Maomao Zhang; Julie S. Di Martino; R. M. Bowman; Nathaniel R. Campbell; Sanjeethan C. Baksh; Theresa Simon-Vermot; Isabella S. Kim; Pearce Haldeman; Chandrani Mondal; Vladimir Yong-Gonzales; et al.Mohsen Abu-AkeelTaha MerghoubDrew R. JonesXiphias Ge ZhuArshi AroraCharlotte AriyanKivanç BirsoyJedd D. WolchokKatherine S. PanageasTravis HollmannJose Javier Bravo-CorderoRichard M. White Adipocyte-Derived Lipids Mediate Melanoma Progression via FATP Proteins. Cancer Discovery 2018, 8, 1006-1025, 10.1158/2159-8290.cd-17-1371.
- Ikrame Lazar; Emily Clement; Stéphanie Dauvillier; Delphine Milhas; Manuelle Ducoux-Petit; Sophie Legonidec; Cédric Moro; Vanessa Soldan; Stéphane Dalle; Stéphanie Balor; et al.Muriel GolzioOdile Burlet-SchiltzPhilippe ValetCatherine MullerLaurence Nieto Adipocyte Exosomes Promote Melanoma Aggressiveness through Fatty Acid Oxidation: A Novel Mechanism Linking Obesity and Cancer. Cancer Research 2016, 76, 4051-4057, 10.1158/0008-5472.can-16-0651.
- Hiu Yee Kwan; Xiuqiong Fu; Bin Liu; Xiaojuan Chao; Chi Leung Chan; Huihui Cao; Tao Su; Anfernee Kai Wing Tse; Wang Fun Fong; Zhi-Ling Yu; et al. Subcutaneous Adipocytes Promote Melanoma Cell Growth by Activating the Akt Signaling Pathway. Journal of Biological Chemistry 2014, 289, 30525-30537, 10.1074/jbc.m114.593210.
- Gretchen M. Alicea; Vito W. Rebecca; Aaron R. Goldman; Mitchell E. Fane; Stephen M. Douglass; Reeti Behera; Marie R. Webster; Curtis H. Kugel; Brett L. Ecker; M. Cecilia Caino; et al.Andrew V. KossenkovHsin-Yao TangDennie T. FrederickKeith T. FlahertyXiaowei XuQin LiuDmitry I. GabrilovichMeenhard HerlynIan A. BlairZachary T. SchugDavid W. SpeicherAshani T. Weeraratna Changes in Aged Fibroblast Lipid Metabolism Induce Age-Dependent Melanoma Cell Resistance to Targeted Therapy via the Fatty Acid Transporter FATP2. Cancer Discovery 2020, 10, 1282-1295, 10.1158/2159-8290.cd-20-0329.
- Yasufumi Goto; Yuriko Matsuzaki; Sachiko Kurihara; Ayako Shimizu; Tsutomu Okada; Kazuhiko Yamamoto; Hiroshi Murata; Minoru Takata; Hiroyuki Aburatani; Dave S.B. Hoon; et al.Toshiaki SaidaYutaka Kawakami A New Melanoma Antigen Fatty Acid-Binding Protein 7, Involved in Proliferation and Invasion, Is a Potential Target for Immunotherapy and Molecular Target Therapy. Cancer Research 2006, 66, 4443-4449, 10.1158/0008-5472.can-05-2505.
- Ana Slipicevic; Kjersti Jørgensen; Martina Skrede; Anne Katrine Ree Rosnes; Gunhild Trøen; Ben Edavidson; Vivi Ann Florenes; The fatty acid binding protein 7 (FABP7) is involved in proliferation and invasion of melanoma cells. BMC Cancer 2008, 8, 276-276, 10.1186/1471-2407-8-276.
- Yasufumi Goto; Kazuo Koyanagi; Norihiko Narita; Yutaka Kawakami; Minoru Takata; Aya Uchiyama; Linhda Nguyen; Tung Nguyen; Xing Ye; Donald L. Morton; et al.Dave S.B. Hoon Aberrant Fatty Acid-Binding Protein-7 Gene Expression in Cutaneous Malignant Melanoma. Journal of Investigative Dermatology 2010, 130, 221-229, 10.1038/jid.2009.195.
- Gloria Pascual; Alexandra Avgustinova; Stefania Mejetta; Mercè Martín; Andrés Castellanos; Camille Stephan-Otto Attolini; Antoni Berenguer; Neus Prats; Agustí Toll; Juan Antonio Hueto; et al.Juan Antonio Hueto Coro BescósStefania Mejetta Luciano Di CroceGloria Pascual Alexandra Avgustinova Mercè Martín Andrés Castellanos Camille Stephan-Otto Attolini Antoni Berenguer Neus Prats Salvador Aznar Benitah Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 2016, 541, 41-45, 10.1038/nature20791.
- Claudia Wellbrock; Imanol Arozarena; Microphthalmia‐associated transcription factor in melanoma development and MAP ‐kinase pathway targeted therapy. Pigment Cell & Melanoma Research 2015, 28, 390-406, 10.1111/pcmr.12370.
- Erez Feige; Satoru Yokoyama; Carmit Levy; Mehdi Khaled; Vivien Igras; Richard J. Lin; Stephen Lee; Hans R. Widlund; Scott R. Granter; Andrew L. Kung; et al.David E. Fisher Hypoxia-induced transcriptional repression of the melanoma-associated oncogene MITF. Proceedings of the National Academy of Sciences 2011, 108, E924-E933, 10.1073/pnas.1106351108.
- Yann Cheli; Sandy Giuliano; Nina Fenouille; Maryline Allegra; Veronique Hofman; Paul Hofman; P Bahadoran; J-P Lacour; Sophie Tartaredeckert; Corine Bertolotto; et al.Robert Ballotti Hypoxia and MITF control metastatic behaviour in mouse and human melanoma cells. Oncogene 2011, 31, 2461-2470, 10.1038/onc.2011.425.
- Pakavarin Louphrasitthiphol; Ioanna Ledaki; Jagat Chauhan; Paola Falletta; Robert Siddaway; Francesca M. Buffa; David R. Mole; Tomoyoshi Soga; Colin R. Goding; MITF controls the TCA cycle to modulate the melanoma hypoxia response. Pigment Cell & Melanoma Research 2019, 32, 792-808, 10.1111/pcmr.12802.
- Jennifer Ferguson; Michael P. Smith; Isabel Zudaire; Claudia Wellbrock; Imanol Arozarena; Glucose availability controls ATF4-mediated MITF suppression to drive melanoma cell growth. Oncotarget 2017, 8, 32946-32959, 10.18632/oncotarget.16514.
- Jennifer Landsberg; Judith Kohlmeyer; Marcel Renn; Tobias Bald; Meri Rogava; Mira Cron; Martina Fatho; Volker Lennerz; Thomas Wölfel; Michael Hölzel; et al.Thomas Tüting Melanomas resist T-cell therapy through inflammation-induced reversible dedifferentiation. Nature 2012, 490, 412-416, 10.1038/nature11538.
- Stefanie Riesenberg; Angela Groetchen; Robert Siddaway; Tobias Bald; Julia Reinhardt; Denise Smorra; Judith Kohlmeyer; Marcel Renn; Bengt Phung; Pia Aymans; et al.Tobias SchmidtVeit HornungIrwin DavidsonColin R. GodingGöran B JönssonJennifer LandsbergThomas TütingMichael Hölzel MITF and c-Jun antagonism interconnects melanoma dedifferentiation with pro-inflammatory cytokine responsiveness and myeloid cell recruitment. Nature Communications 2015, 6, 8755, 10.1038/ncomms9755.
- Paola Falletta; Luis Sanchez-Del-Campo; Jagat Chauhan; Maike Effern; Amy Kenyon; Christopher J. Kershaw; Robert Siddaway; Richard J. Lisle; Rasmus Freter; Matthew J. Daniels; et al.Xin LuThomas TütingMark MiddletonFrancesca M. BuffaAnne E. WillisGraham D PavittZe'ev A. RonaiTatjana Sauka-SpenglerMichael HölzelColin R. Goding Translation reprogramming is an evolutionarily conserved driver of phenotypic plasticity and therapeutic resistance in melanoma. Genes & Development 2017, 31, 18-33, 10.1101/gad.290940.116.
- Keith S. Hoek; Natalie C. Schlegel; Patricia Brafford; Antje Sucker; Selma Ugurel; Rajiv Kumar; Barbara L. Weber; Katherine L. Nathanson; David J. Phillips; Meenhard Herlyn; et al.Dirk SchadendorfReinhard Dummer Metastatic potential of melanomas defined by specific gene expression profiles with no BRAF signature. Pigment Cell Research 2006, 19, 290-302, 10.1111/j.1600-0749.2006.00322.x.
- Jennifer Tsoi; Lidia Robert; Kim Paraiso; Carlos Galvan; Katherine M. Sheu; Johnson Lay; Deborah J.L. Wong; Mohammad Atefi; Roksana Shirazi; Xiaoyan Wang; et al.Daniel BraasCatherine S. GrassoNicolaos PalaskasAntoni RibasThomas G. Graeber Multi-stage Differentiation Defines Melanoma Subtypes with Differential Vulnerability to Drug-Induced Iron-Dependent Oxidative Stress. Cancer Cell 2018, 33, 890-904.e5, 10.1016/j.ccell.2018.03.017.
- Florian Rambow; Aljosja Rogiers; Oskar Marin-Bejar; Sara Aibar; Julia Femel; Michael Dewaele; Panagiotis Karras; Daniel Brown; Young Hwan Chang; Maria Debiec-Rychter; et al.Carmen AdriaensEnrico RadaelliPascal WolterOliver BechterReinhard DummerMitchell LevesqueAdriano PirisDennie T. FrederickGenevieve M. BolandKeith T. FlahertyJoost Van Den OordThierry VoetStein AertsAmanda W. LundJean-Christophe Marine Toward Minimal Residual Disease-Directed Therapy in Melanoma. Cell 2018, 174, 843-855.e19, 10.1016/j.cell.2018.06.025.
- Keith S. Hoek; Ossia M Eichhoff; Natalie C. Schlegel; Udo Döbbeling; Nikita Kobert; Leo Schaerer; Silvio Hemmi; Reinhard Dummer; In vivo Switching of Human Melanoma Cells between Proliferative and Invasive States. Cancer Research 2008, 68, 650-656, 10.1158/0008-5472.can-07-2491.
- Julie Caramel; Eftychios Papadogeorgakis; Louise Hill; Gareth J. Browne; Geoffrey Richard; Anne Wierinckx; Gerald Saldanha; Joy Osborne; Peter Hutchinson; Gina Tse; et al.Joël LachuerAlain PuisieuxJ Howard PringleStéphane AnsieauEugene Tulchinsky A Switch in the Expression of Embryonic EMT-Inducers Drives the Development of Malignant Melanoma. Cancer Cell 2013, 24, 466-480, 10.1016/j.ccr.2013.08.018.
- Yurena Vivas-García; Paola Falletta; Jana Liebing; Pakavarin Louphrasitthiphol; YongMei Feng; Jagat Chauhan; David A. Scott; Nicole Glodde; Ana Chocarro-Calvo; Sarah Bonham; et al.Andrei L. OstermanRoman FischerZe’Ev RonaiCustodia García-JiménezMichael HölzelColin R. Goding Lineage-Restricted Regulation of SCD and Fatty Acid Saturation by MITF Controls Melanoma Phenotypic Plasticity. Molecular Cell 2020, 77, 120-137.e9, 10.1016/j.molcel.2019.10.014.


