 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Franco Trevisani | -- | 2701 | 2022-10-17 20:27:59 | | | |
| 2 | Peter Tang | Meta information modification | 2701 | 2022-10-18 03:17:54 | | |
Video Upload Options
Hepatocellular carcinoma (HCC) is the most common liver cancer and a relevant global health problem. Immune checkpoint inhibitors (ICIs) represent the most effective systemic treatment for HCC. However, due to primary resistance, approximately 40% of HCC patients do not achieve a disease control with ICIs. Moreover, a similar proportion will experience disease progression after an initial response caused by secondary resistance.
1. Introduction
2. Tumour Immunity Cycle
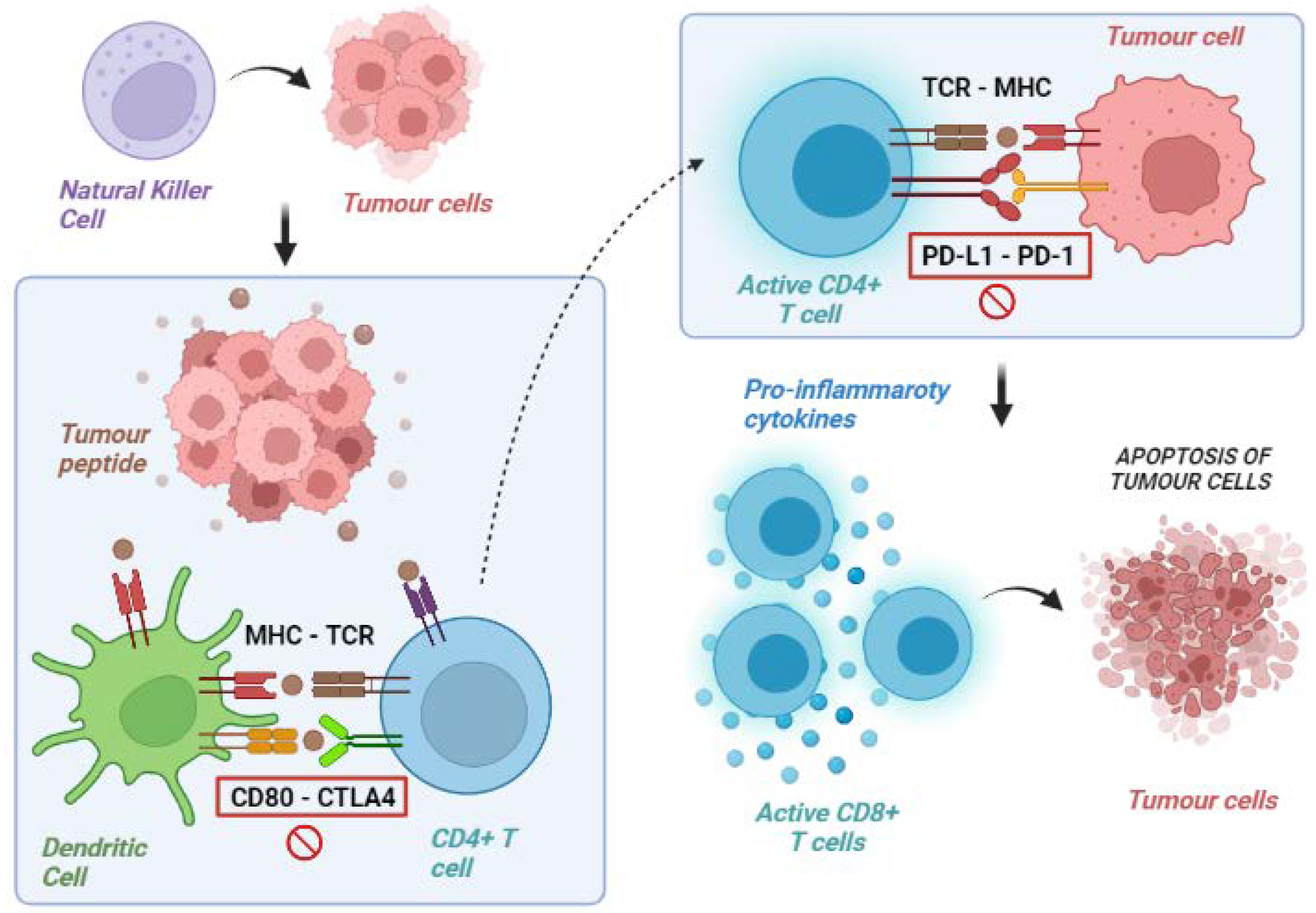
3. Tumour Immune Escape
|
CTLA-4 |
PD-1 |
PD-L1 |
LAG-3 |
|---|---|---|---|
|
Ipilimumab |
Nivolumab |
Durvalumab |
Relatlimab |
|
Tremelimumab |
Pembrolizumab |
Avelumab |
|
|
Camrelizumab |
Atezolizumab |
||
|
Dostarlimab |
|||
|
Toripalimab |
|||
|
Spartalizumab |
|||
|
Cempilimab |
|||
|
Sintilimab |
|||
|
Serpulimab |
|||
|
Nofazinlimab |
|||
|
Penpulimab |
CTLA-4: cytotoxic T lymphocyte-associated protein 4; PD-1: programmed death-1; PD-L1: programmed death 1-ligand; LAG3: lymphocyte activation gene-3.
4. Mechanisms of Primary ICI Resistance
4.1. Tumour-Intrinsic Mechanisms
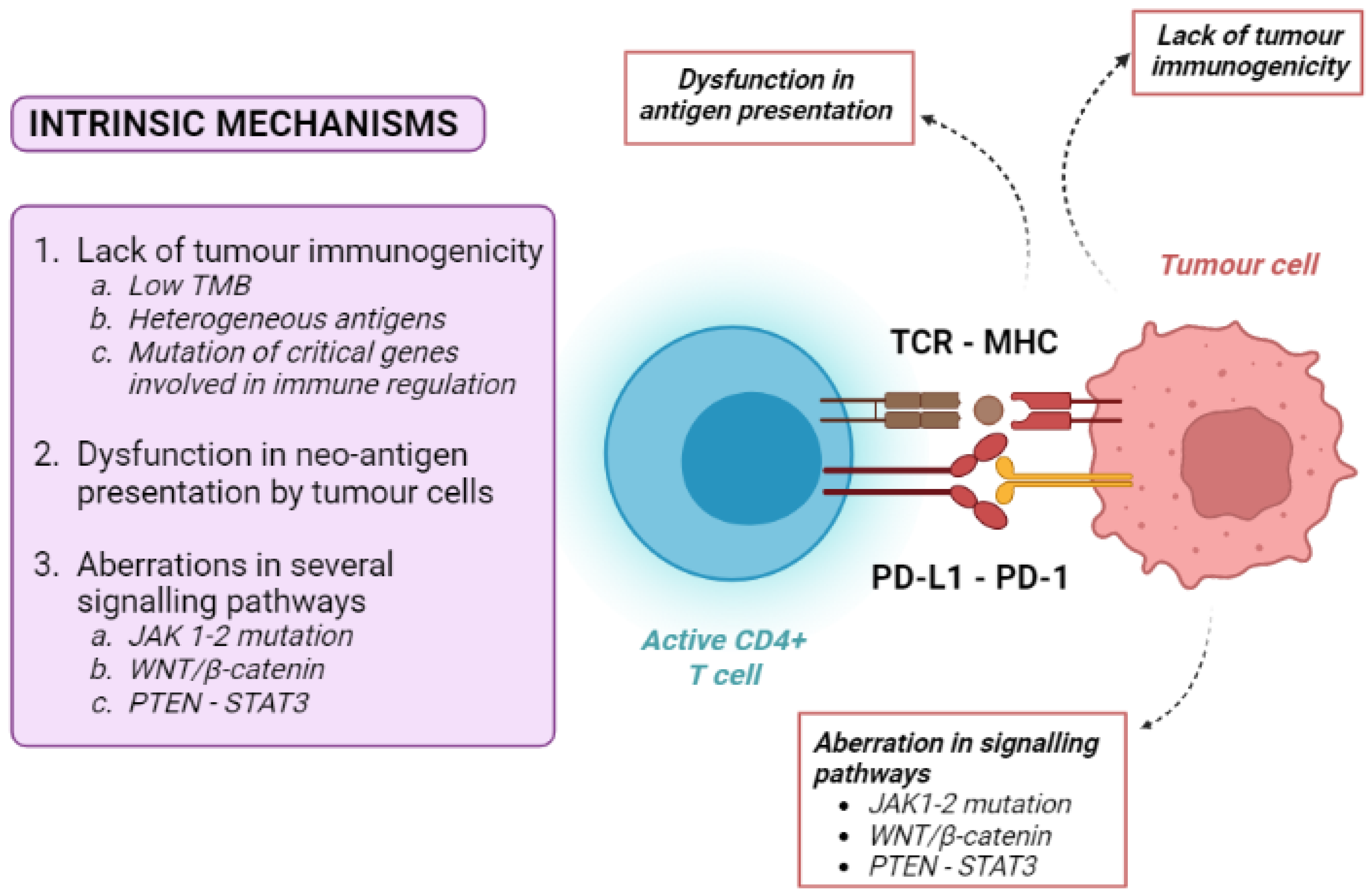
4.2. Tumour-Extrinsic Mechanisms
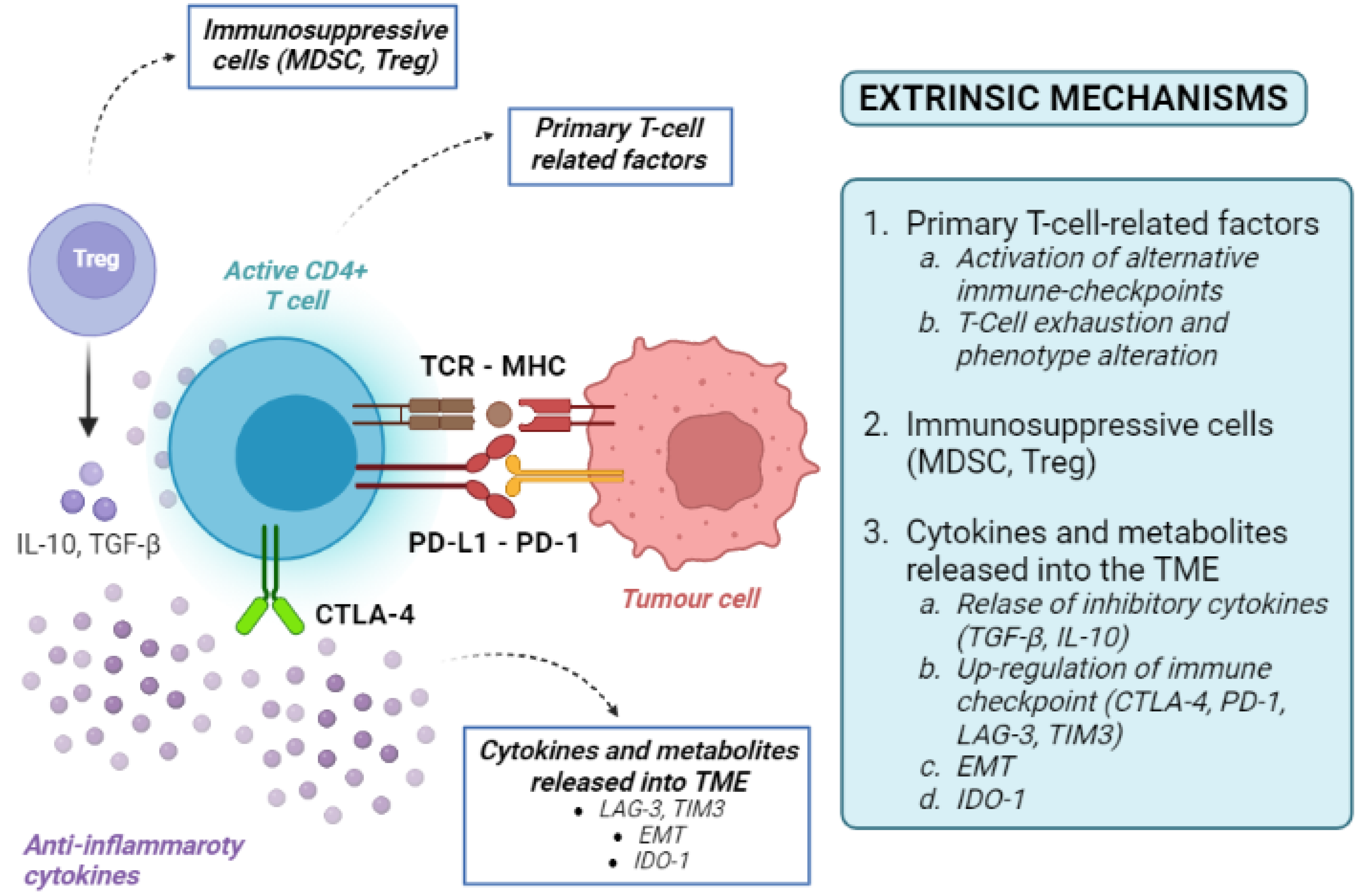
5. Mechanisms of Acquired ICI Resistance
6. Current Strategies in Immunotherapy for HCC
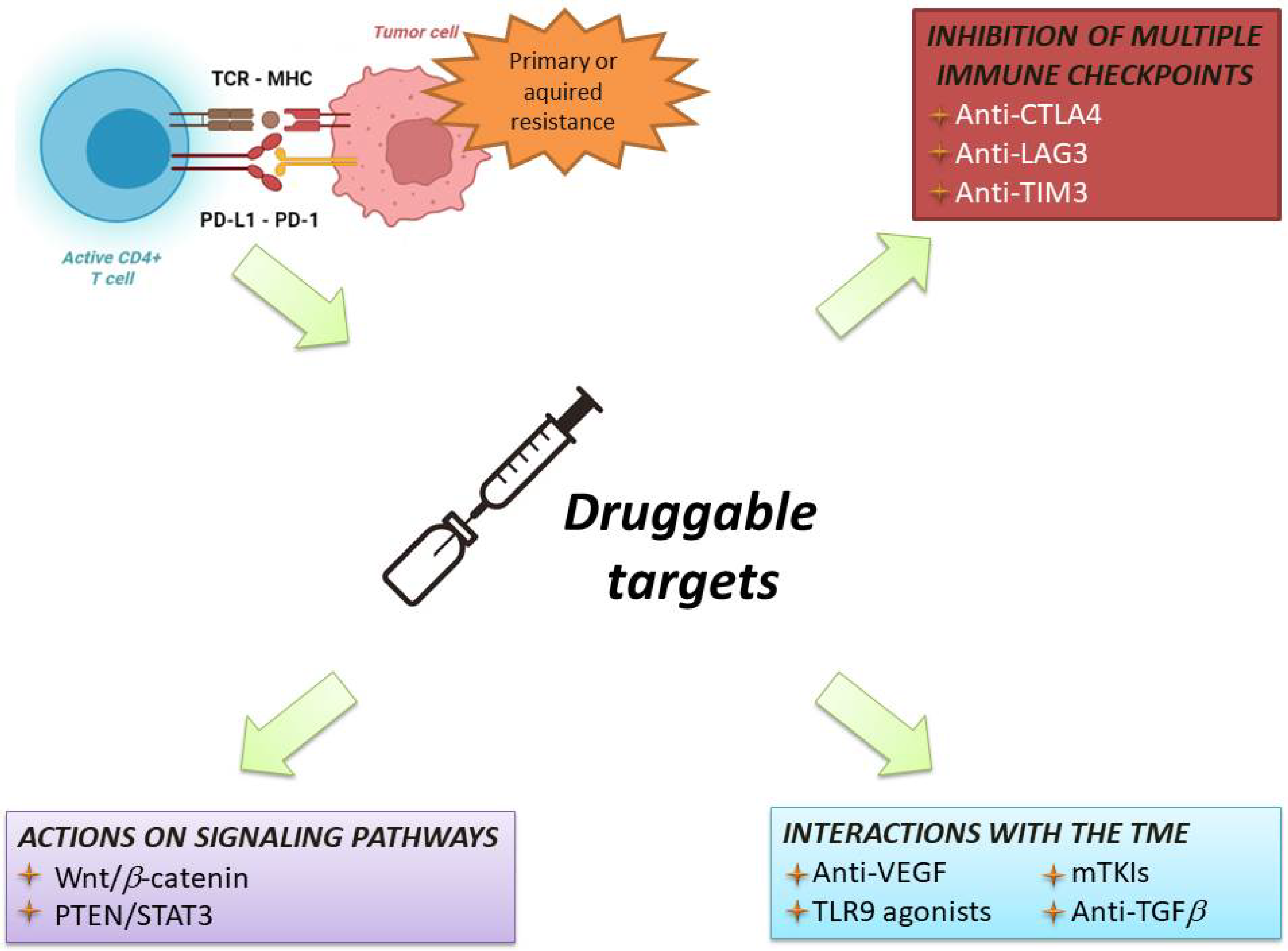
References
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune Checkpoint Blockade: A Common Denominator Approach to Cancer Therapy. Cancer Cell 2015, 27, 450–461.
- Sharma, P.; Allison, J.P. Immune Checkpoint Targeting in Cancer Therapy: Toward Combination Strategies with Curative Potential. Cell 2015, 161, 205–214.
- Rumgay, H.; Ferlay, J.; De Martel, C.; Georges, D.; Ibrahim, A.S.; Zheng, R.; Wei, W.; Lemmens, V.E.P.P.; Soerjomataram, I. Global, Regional and National Burden of Primary Liver Cancer by Subtype. Eur. J. Cancer 2022, 161, 108–118.
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905.
- Abou-Alfa Ghassan, K.; Lau, G.; Kudo, M.; Chan, S.L.; Kelley, R.K.; Furuse, J.; Sukeepaisarnjaroen, W.; Kang, Y.-K.; Van Dao, T.; De Toni, E.N.; et al. Tremelimumab plus Durvalumab in Unresectable Hepatocellular Carcinoma. NEJM Evid. 2022, 1, EVIDoa2100070.
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.-F.; De Oliveira, A.C.; Santoro, A.; Raoul, J.-L.; Forner, A.; et al. Sorafenib in Advanced Hepatocellular Carcinoma. N. Engl. J. Med. 2008, 359, 378–390.
- Woo, S.-R.; Turnis, M.E.; Goldberg, M.V.; Bankoti, J.; Selby, M.; Nirschl, C.J.; Bettini, M.L.; Gravano, D.M.; Vogel, P.; Liu, C.L.; et al. Immune Inhibitory Molecules LAG-3 and PD-1 Synergistically Regulate T-Cell Function to Promote Tumoral Immune Escape. Cancer Res. 2012, 72, 917–927.
- Benci, J.L.; Xu, B.; Qiu, Y.; Wu, T.J.; Dada, H.; Twyman-Saint Victor, C.; Cucolo, L.; Lee, D.S.M.; Pauken, K.E.; Huang, A.C.; et al. Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell 2016, 167, 1540–1554.
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H.; et al. Adaptive Resistance to Therapeutic PD-1 Blockade Is Associated with Upregulation of Alternative Immune Checkpoints. Nat. Commun. 2016, 7, 10501.
- Shin, D.S.; Zaretsky, J.M.; Escuin-Ordinas, H.; Garcia-Diaz, A.; Hu-Lieskovan, S.; Kalbasi, A.; Grasso, C.S.; Hugo, W.; Sandoval, S.; Torrejon, D.Y.; et al. Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discov. 2017, 7, 188–201.
- Chen, D.S.; Mellman, I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013, 39, 1–10.
- Chen, D.S.; Irving, B.A.; Hodi, F.S. Molecular Pathways: Next-Generation Immunotherapy—Inhibiting Programmed Death-Ligand 1 and Programmed Death-1. Clin. Cancer Res. 2012, 18, 6580–6587.
- McGranahan, N.; Furness, A.J.S.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal Neoantigens Elicit T Cell Immunoreactivity and Sensitivity to Immune Checkpoint Blockade. Science 2016, 351, 1463–1469.
- Coulie, P.G.; Van den Eynde, B.J.; Van der Bruggen, P.; Boon, T. Tumour Antigens Recognized by T Lymphocytes: At the Core of Cancer Immunotherapy. Nat. Rev. Cancer 2014, 14, 135–146.
- Zhang, Q.; Jia, Q.; Zhang, J.; Zhu, B. Neoantigens in Precision Cancer Immunotherapy: From Identification to Clinical Applications. Chin. Med. J. 2022.
- Chambers, C.A.; Kuhns, M.S.; Egen, J.G.; Allison, J.P. CTLA-4-Mediated Inhibition in Regulation of T Cell Responses: Mechanisms and Manipulation in Tumor Immunotherapy. Annu. Rev. Immunol. 2001, 19, 565–594.
- Liechtenstein, T.; Dufait, I.; Bricogne, C.; Lanna, A.; Pen, J.; Breckpot, K.; Escors, D. PD-L1/PD-1 Co-Stimulation, a Brake for T Cell Activation and a T Cell Differentiation Signal. J. Clin. Cell Immunol. 2012, S12, 006.
- Graydon, C.G.; Mohideen, S.; Fowke, K.R. LAG3’s Enigmatic Mechanism of Action. Front. Immunol. 2021, 11, 615317.
- Blackburn, S.D.; Shin, H.; Haining, W.N.; Zou, T.; Workman, C.J.; Polley, A.; Betts, M.R.; Freeman, G.J.; Vignali, D.A.A.; Wherry, E.J. Coregulation of CD8+ T Cell Exhaustion during Chronic Viral Infection by Multiple Inhibitory Receptors. Nat. Immunol. 2009, 10, 29–37.
- Abdel-Wahab, N.; Shah, M.; Lopez-Olivo, M.A.; Suarez-Almazor, M.E. Use of Immune Checkpoint Inhibitors in the Treatment of Patients With Cancer and Preexisting Autoimmune Disease: A Systematic Review. Ann. Intern. Med. 2018, 168, 121–130.
- Zou, W.; Wolchok, J.D.; Chen, L. PD-L1 (B7-H1) and PD-1 Pathway Blockade for Cancer Therapy: Mechanisms, Response Biomarkers, and Combinations. Sci. Transl. Med. 2016, 8, 328rv4.
- Ribas, A. Tumor Immunotherapy Directed at PD-1. N. Engl. J. Med. 2012, 366, 2517–2519.
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2017, 168, 542.
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer Immunology. Mutational Landscape Determines Sensitivity to PD-1 Blockade in Non-Small Cell Lung Cancer. Science 2015, 348, 124–128.
- Van Allen, E.M.; Miao, D.; Schilling, B.; Shukla, S.A.; Blank, C.; Zimmer, L.; Sucker, A.; Hillen, U.; Foppen, M.H.G.; Goldinger, S.M.; et al. Genomic Correlates of Response to CTLA-4 Blockade in Metastatic Melanoma. Science 2015, 350, 207–211.
- Chowell, D.; Morris, L.G.T.; Grigg, C.M.; Weber, J.K.; Samstein, R.M.; Makarov, V.; Kuo, F.; Kendall, S.M.; Requena, D.; Riaz, N.; et al. Patient HLA Class I Genotype Influences Cancer Response to Checkpoint Blockade Immunotherapy. Science 2018, 359, 582–587.
- Ichinokawa, K.; Nakanishi, Y.; Hida, Y.; Tsuchikawa, T.; Kato, T.; Itoh, T.; Kaji, M.; Kaga, K.; Hirano, S. Downregulated Expression of Human Leukocyte Antigen Class I Heavy Chain Is Associated with Poor Prognosis in Non-Small-Cell Lung Cancer. Oncol. Lett. 2019, 18, 117–126.
- McGranahan, N.; Rosenthal, R.; Hiley, C.T.; Rowan, A.J.; Watkins, T.B.K.; Wilson, G.A.; Birkbak, N.J.; Veeriah, S.; Van Loo, P.; Herrero, J.; et al. Allele-Specific HLA Loss and Immune Escape in Lung Cancer Evolution. Cell 2017, 171, 1259–1271.
- Li, X.; Xiang, Y.; Li, F.; Yin, C.; Li, B.; Ke, X. WNT/β-Catenin Signaling Pathway Regulating T Cell-Inflammation in the Tumor Microenvironment. Front. Immunol. 2019, 10, 2293.
- Sia, D.; Jiao, Y.; Martinez-Quetglas, I.; Kuchuk, O.; Villacorta-Martin, C.; Castro de Moura, M.; Putra, J.; Camprecios, G.; Bassaganyas, L.; Akers, N.; et al. Identification of an Immune-Specific Class of Hepatocellular Carcinoma, Based on Molecular Features. Gastroenterology 2017, 153, 812–826.
- Llovet, J.M.; Montal, R.; Sia, D.; Finn, R.S. Molecular Therapies and Precision Medicine for Hepatocellular Carcinoma. Nat. Rev. Clin. Oncol. 2018, 15, 599–616.
- Harding, J.J.; Nandakumar, S.; Armenia, J.; Khalil, D.N.; Albano, M.; Ly, M.; Shia, J.; Hechtman, J.F.; Kundra, R.; El Dika, I.; et al. Prospective Genotyping of Hepatocellular Carcinoma: Clinical Implications of Next-Generation Sequencing for Matching Patients to Targeted and Immune Therapies. Clin. Cancer Res. 2019, 25, 2116–2126.
- Pinyol, R.; Sia, D.; Llovet, J.M. Immune Exclusion-Wnt/CTNNB1 Class Predicts Resistance to Immunotherapies in HCC. Clin. Cancer Res. 2019, 25, 2021–2023.
- Jung, K.H.; Yoo, W.; Stevenson, H.L.; Deshpande, D.; Shen, H.; Gagea, M.; Yoo, S.-Y.; Wang, J.; Eckols, T.K.; Bharadwaj, U.; et al. Multifunctional Effects of a Small-Molecule STAT3 Inhibitor on NASH and Hepatocellular Carcinoma in Mice. Clin. Cancer Res. 2017, 23, 5537–5546.
- Tsimberidou, A.M.; De Achaval, S.; Alibhai, I.; Kaseb, A.O. First-in-Man Phase I Clinical Trial Evaluating TTI-101, an Orally Bioavailable, Small Molecule Inhibitor of STAT3, in Patients with Advanced Solid Tumors. J. Clin. Oncol. 2021, 39, TPS3158.
- Joyce, J.A.; Fearon, D.T. T Cell Exclusion, Immune Privilege, and the Tumor Microenvironment. Science 2015, 348, 74–80.
- Wang, F.; Wang, S.; Zhou, Q. The Resistance Mechanisms of Lung Cancer Immunotherapy. Front. Oncol. 2020, 10, 568059.
- Matsuda, Y.; Yamagiwa, Y.; Fukushima, K.; Ueno, Y.; Shimosegawa, T. Expression of Galectin-3 Involved in Prognosis of Patients with Hepatocellular Carcinoma. Hepatol. Res. 2008, 38, 1098–1111.
- Yan, W.; Liu, X.; Ma, H.; Zhang, H.; Song, X.; Gao, L.; Liang, X.; Ma, C. Tim-3 Fosters HCC Development by Enhancing TGF-β-Mediated Alternative Activation of Macrophages. Gut 2015, 64, 1593–1604.
- Yarchoan, M.; Xing, D.; Luan, L.; Xu, H.; Sharma, R.B.; Popovic, A.; Pawlik, T.M.; Kim, A.K.; Zhu, Q.; Jaffee, E.M.; et al. Characterization of the Immune Microenvironment in Hepatocellular Carcinoma. Clin. Cancer Res. 2017, 23, 7333–7339.
- Kalluri, R.; Weinberg, R.A. The Basics of Epithelial-Mesenchymal Transition. J. Clin. Investig. 2009, 119, 1420–1428.
- Terry, S.; Savagner, P.; Ortiz-Cuaran, S.; Mahjoubi, L.; Saintigny, P.; Thiery, J.-P.; Chouaib, S. New Insights into the Role of EMT in Tumor Immune Escape. Mol. Oncol. 2017, 11, 824–846.
- Soundararajan, R.; Fradette, J.J.; Konen, J.M.; Moulder, S.; Zhang, X.; Gibbons, D.L.; Varadarajan, N.; Wistuba, I.I.; Tripathy, D.; Bernatchez, C.; et al. Targeting the Interplay between Epithelial-to-Mesenchymal-Transition and the Immune System for Effective Immunotherapy. Cancers 2019, 11, 714.
- Ye, L.-Y.; Chen, W.; Bai, X.-L.; Xu, X.-Y.; Zhang, Q.; Xia, X.-F.; Sun, X.; Li, G.-G.; Hu, Q.-D.; Fu, Q.-H.; et al. Hypoxia-Induced Epithelial-to-Mesenchymal Transition in Hepatocellular Carcinoma Induces an Immunosuppressive Tumor Microenvironment to Promote Metastasis. Cancer Res. 2016, 76, 818–830.
- Kim, J.M.; Chen, D.S. Immune Escape to PD-L1/PD-1 Blockade: Seven Steps to Success (or Failure). Ann. Oncol. 2016, 27, 1492–1504.
- Nakao, A.; Afrakhte, M.; Morén, A.; Nakayama, T.; Christian, J.L.; Heuchel, R.; Itoh, S.; Kawabata, M.; Heldin, N.E.; Heldin, C.H.; et al. Identification of Smad7, a TGFbeta-Inducible Antagonist of TGF-Beta Signalling. Nature 1997, 389, 631–635.
- Massagué, J. TGFβ Signalling in Context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630.
- Blobe, G.C.; Schiemann, W.P.; Lodish, H.F. Role of Transforming Growth Factor Beta in Human Disease. N. Engl. J. Med. 2000, 342, 1350–1358.
- Eisenstein, E.M.; Williams, C.B. The T(Reg)/Th17 Cell Balance: A New Paradigm for Autoimmunity. Pediatr. Res. 2009, 65, 26R–31R.
- Roes, J.; Choi, B.K.; Cazac, B.B. Redirection of B Cell Responsiveness by Transforming Growth Factor Beta Receptor. Proc. Natl. Acad. Sci. USA 2003, 100, 7241–7246.
- Chang, C.-H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; Van der Windt, G.J.W.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241.
- Sangaletti, S.; Tripodo, C.; Santangelo, A.; Castioni, N.; Portararo, P.; Gulino, A.; Botti, L.; Parenza, M.; Cappetti, B.; Orlandi, R.; et al. Mesenchymal Transition of High-Grade Breast Carcinomas Depends on Extracellular Matrix Control of Myeloid Suppressor Cell Activity. Cell Rep. 2016, 17, 233–248.
- Audrito, V.; Managò, A.; Gaudino, F.; Sorci, L.; Messana, V.G.; Raffaelli, N.; Deaglio, S. NAD-Biosynthetic and Consuming Enzymes as Central Players of Metabolic Regulation of Innate and Adaptive Immune Responses in Cancer. Front. Immunol. 2019, 10, 1720.
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in Cancer Immunotherapy. Science 2015, 348, 69–74.
- Kato, S.; Goodman, A.; Walavalkar, V.; Barkauskas, D.A.; Sharabi, A.; Kurzrock, R. Hyperprogressors after Immunotherapy: Analysis of Genomic Alterations Associated with Accelerated Growth Rate. Clin. Cancer Res. 2017, 23, 4242–4250.
- Gettinger, S.; Choi, J.; Hastings, K.; Truini, A.; Datar, I.; Sowell, R.; Wurtz, A.; Dong, W.; Cai, G.; Melnick, M.A.; et al. Impaired HLA Class I Antigen Processing and Presentation as a Mechanism of Acquired Resistance to Immune Checkpoint Inhibitors in Lung Cancer. Cancer Discov. 2017, 7, 1420–1435.
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829.
- Martin, A.M.; Nirschl, T.R.; Nirschl, C.J.; Francica, B.J.; Kochel, C.M.; Van Bokhoven, A.; Meeker, A.K.; Lucia, M.S.; Anders, R.A.; DeMarzo, A.M.; et al. Paucity of PD-L1 Expression in Prostate Cancer: Innate and Adaptive Immune Resistance. Prostate Cancer Prostatic Dis. 2015, 18, 325–332.
- Tovoli, F.; De Lorenzo, S.; Trevisani, F. Immunotherapy with Checkpoint Inhibitors for Hepatocellular Carcinoma: Where Are We Now? Vaccines 2020, 8, 578.
- Yau, T.; Park, J.-W.; Finn, R.S.; Cheng, A.-L.; Mathurin, P.; Edeline, J.; Kudo, M.; Harding, J.J.; Merle, P.; Rosmorduc, O.; et al. Nivolumab versus Sorafenib in Advanced Hepatocellular Carcinoma (CheckMate 459): A Randomised, Multicentre, Open-Label, Phase 3 Trial. Lancet Oncol. 2022, 23, 77–90.
- Zhu, A.X.; Finn, R.S.; Edeline, J.; Cattan, S.; Ogasawara, S.; Palmer, D.; Verslype, C.; Zagonel, V.; Fartoux, L.; Vogel, A.; et al. Pembrolizumab in Patients with Advanced Hepatocellular Carcinoma Previously Treated with Sorafenib (KEYNOTE-224): A Non-Randomised, Open-Label Phase 2 Trial. Lancet Oncol. 2018, 19, 940–952.
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.-Y.; Choo, S.-P.; Trojan, J.; Welling, T.H.; et al. Nivolumab in Patients with Advanced Hepatocellular Carcinoma (CheckMate 040): An Open-Label, Non-Comparative, Phase 1/2 Dose Escalation and Expansion Trial. Lancet 2017, 389, 2492–2502.

