Hepatocellular carcinoma (HCC) is the most common liver cancer and a relevant global health problem. Immune checkpoint inhibitors (ICIs) represent the most effective systemic treatment for HCC. However, due to primary resistance, approximately 40% of HCC patients do not achieve a disease control with ICIs. Moreover, a similar proportion will experience disease progression after an initial response caused by secondary resistance.
- liver cancer
- hepatocellular carcinoma
- immunotherapy
- immune checkpoint inhibitors
- atezolizumab
- bevacizumab
- tremelimumab
- durvalumab
- tumour microenvironment
- resistance
- cirrhosis
1. Introduction
2. Tumour Immunity Cycle
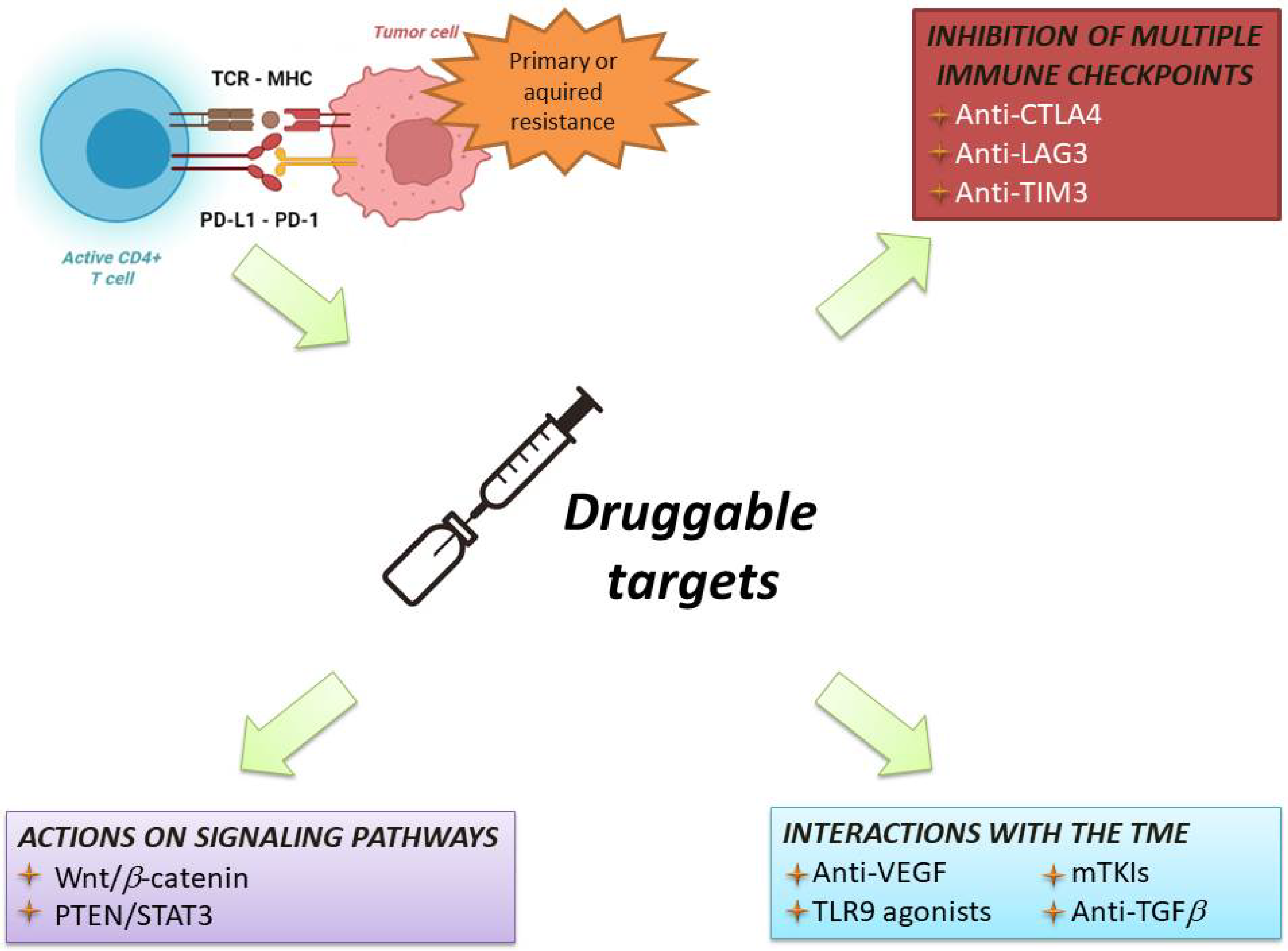
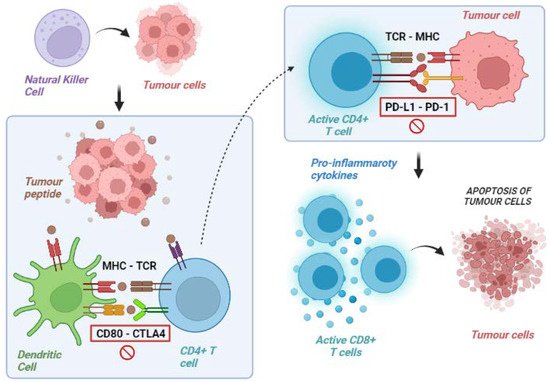
Our immune system is a complex system that operates through two main modalities of response: innate immunity (a non-specific and quick response) and adaptive immunity (subsequently activated antigen-specific response that mediates immune memory) [11]. Although a number of cellular components are involved in this complex system, antitumour immune surveillance is based on T lymphocytes, as demonstrated by several mice models, in which the loss of T-cell activity was associated with uncontrolled tumour growth. T lymphocytes recognise and identify some molecules released from cancer cells as foreign (or “antigens”). These tumour-associated molecules are mutated proteins, showing alterations caused by the high genetic instability of tumours. Indeed, the high degree of genetic diversity in cancer cells accelerates their evolutionary fitness but also increases the divergence from a normal cell, facilitating recognition by the immune system. The frequency of non-synonymous somatic mutations per DNA megabase in the coding genome of a tumour is called “tumour mutational burden” (TMB) [12][13][12,13]. Overall, activating antitumour immunity is a multistep process involving several lymphocyte populations. The uncontrolled growth and the consequent high TMB of cancer cells firstly result in the activation of innate immunity, including natural killer (NK) cells, which target cancer cells and favour their apoptosis leading to the release of tumour-associated antigens. These molecules are released into the tumour microenvironment (TME) and are subsequently recognised by antigen-presenting cells (APCs), which capture these neo-antigens and present them to the corresponding T-cell receptor (TCR) of naïve T lymphocytes via the major histocompatibility complex (MHC). The final step of these immune events is the release of co-stimulatory cytokines by CD4+ T cells and the activation of antigen-specific CD8+ T cells leading to the lymphocyte-mediated destruction of tumour cells (Figure 1) [14][15][14,15].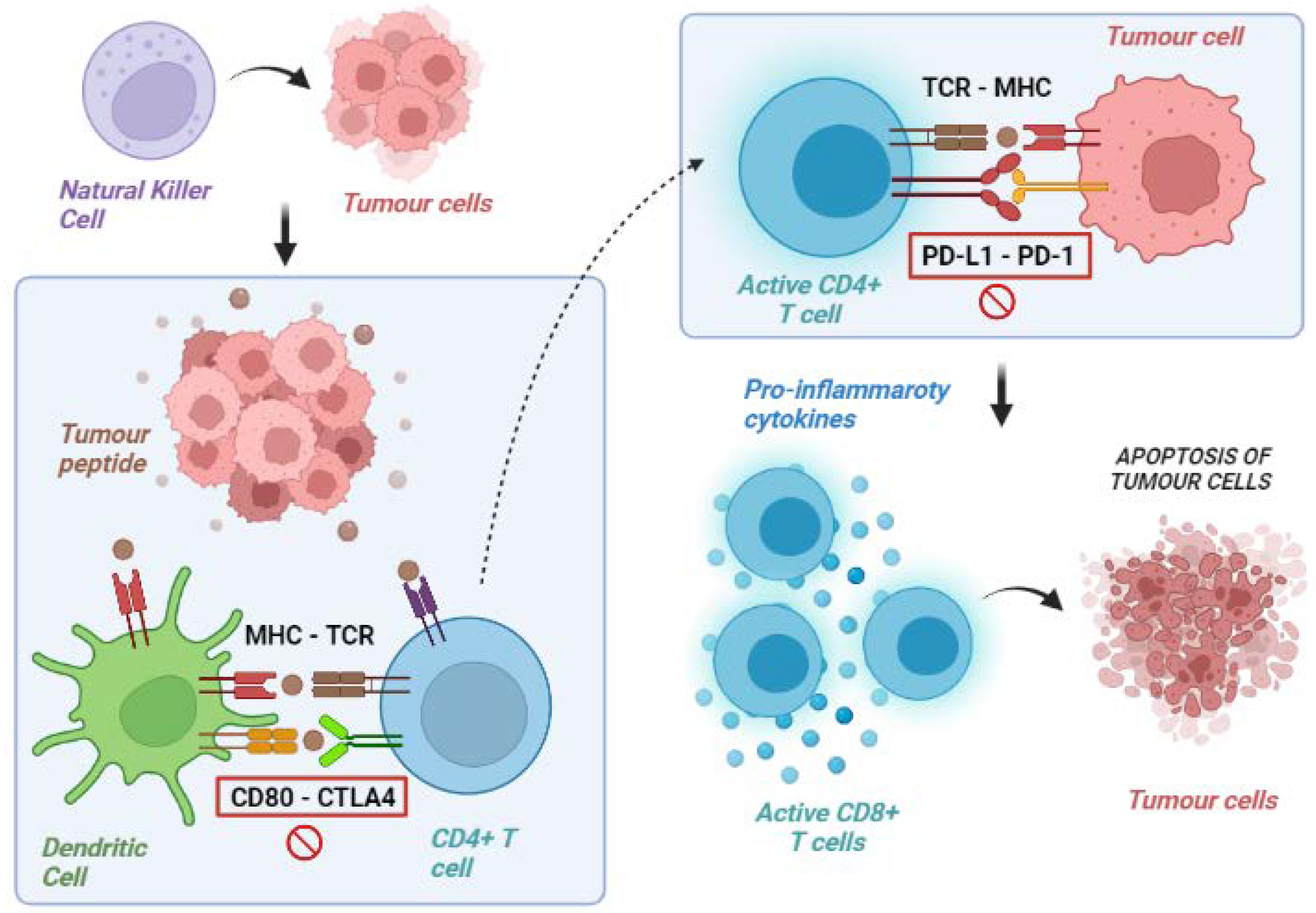
3. Tumour Immune Escape
In acute infections, T cells can eliminate the pathogen. Instead, in the case of advanced tumours, the neo-antigen is rarely eliminated and, consequently, a chronic inflammatory stimulation is established with the possibility of losing effectiveness. In fact, chronic immune stimulation activates negative feedback mechanisms (in particular, the activation of immunosuppressive cells that silence the immune response) and promotes a progressive loss of the cytotoxic capacities of T cells. On the other hand, cancer cells can develop a series of resistance mechanisms aimed at evading the host’s immune control. Namely, tumour cells can acquire the ability to express molecules that inhibit the immune response or reduce the expression of the MHC complex in APCs [16][17][16,17]. The most known immune checkpoint molecules are CTLA4, PD-1 and its ligand PD-L1, and lymphocyte-activation gene 3 (LAG-3). All of them are receptors expressed on the membrane of T cells in different phases. They counterbalance lymphocyte activation in physiological conditions to avoid unnecessary tissue damage, chronic inflammation, and uncontrolled lymphocytic proliferation. In detail, CTLA-4 is expressed by T cells in the priming phase, exerting inhibitory actions following interaction with its ligands, CD80 (B7-1) and CD86 (B7-2), which are expressed on the surface of the APCs. PD-1, instead, is expressed in the effector phase, especially under conditions of chronic exposure to the antigen, and exerts an inhibitory action on the lymphocytes via interaction with its ligands, PD-L1 and PD-L2, found in the TME. Similarly, LAG3 is expressed in lymphocytes during the effector phase [18]. LAG3’s most important ligand is represented by the MHC class II molecules expressed on the surface of APCs, with an interaction ultimately leading to T-cell exhaustion [19]. Tumours can exploit these mechanisms to avoid immune-mediated destruction not only by expressing ligands activating the immune checkpoints themselves but also favouring the development of a tolerant TME through the recruitment of non-neoplastic cells expressing these ligands. Immune checkpoint inhibitors (ICI) are monoclonal antibodies specifically designed to disrupt these ligand/receptor interactions, removing the inhibition of T cells and promoting their antitumoral cytotoxic activity (Table 1) [20][21][22][20,21,22].|
CTLA-4 |
PD-1 |
PD-L1 |
LAG-3 |
|---|---|---|---|
|
Ipilimumab |
Nivolumab |
Durvalumab |
Relatlimab |
|
Tremelimumab |
Pembrolizumab |
Avelumab |
|
|
Camrelizumab |
Atezolizumab |
||
|
Dostarlimab |
|||
|
Toripalimab |
|||
|
Spartalizumab |
|||
|
Cempilimab |
|||
|
Sintilimab |
|||
|
Serpulimab |
|||
|
Nofazinlimab |
|||
|
Penpulimab |
CTLA-4: cytotoxic T lymphocyte-associated protein 4; PD-1: programmed death-1; PD-L1: programmed death 1-ligand; LAG3: lymphocyte activation gene-3.
4. Mechanisms of Primary ICI Resistance
4.1. Tumour-Intrinsic Mechanisms
Tumour intrinsic mechanisms include a lack of tumour immunogenicity (low TMB, heterogeneous antigens, mutation of critical genes involved in immune regulation), defective antigen presentation, and aberrations in several signalling pathways (Figure 2).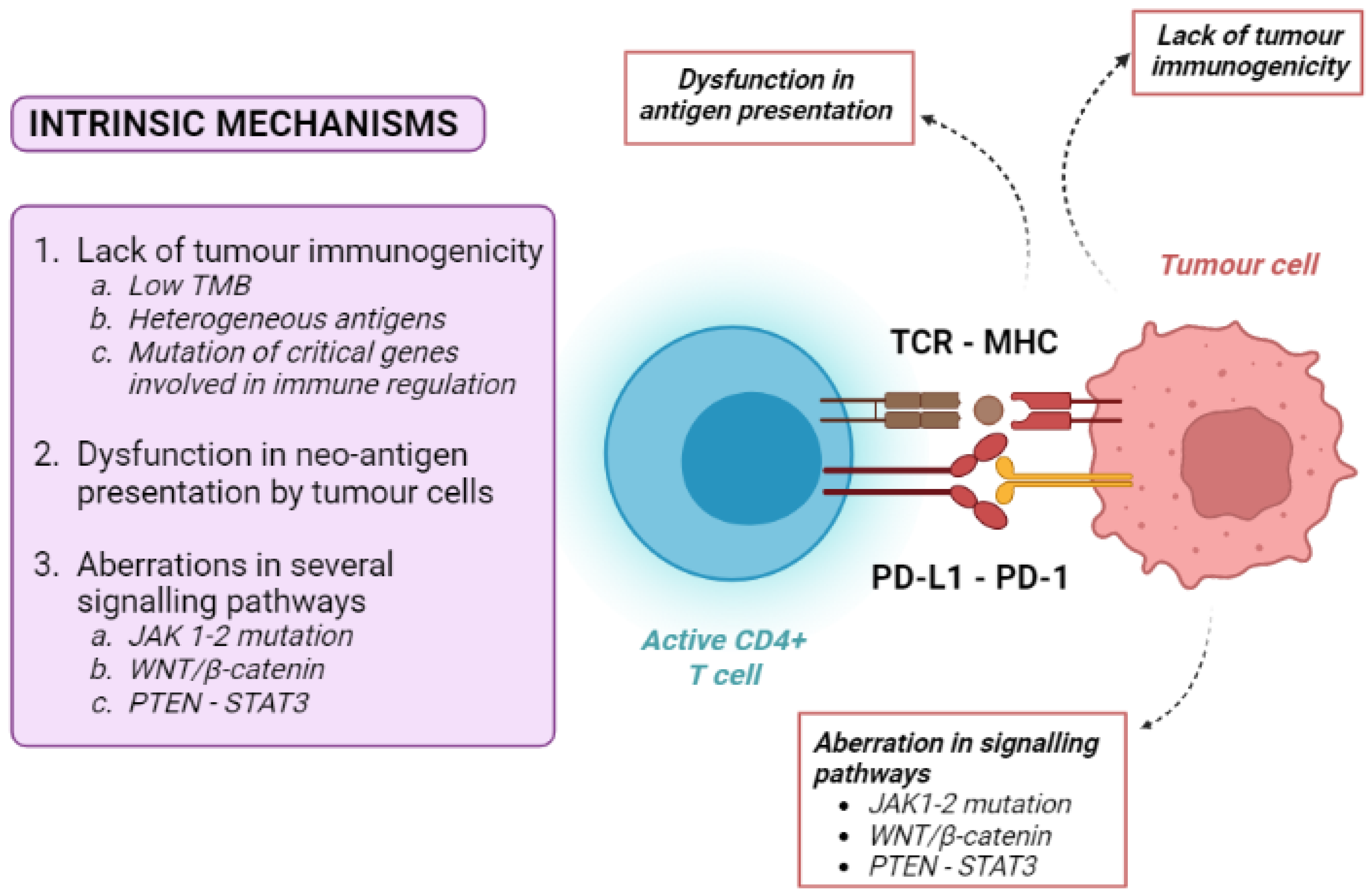
4.2. Tumour-Extrinsic Mechanisms
Tumour-extrinsic mechanisms are mainly due to the immune-suppressive effect of TME. They include primary T-cell-related factors (activation of alternative immune checkpoints, T-cell exhaustion and phenotype alteration), immunosuppressive cells, cytokines and metabolites released into the TME (Figure 3). As the TME includes immune cells, blood vessels, fibroblasts, signalling molecules, and the extracellular matrix surrounding tumour cells, many factors can contribute to the development of an immune-suppressive TME [36].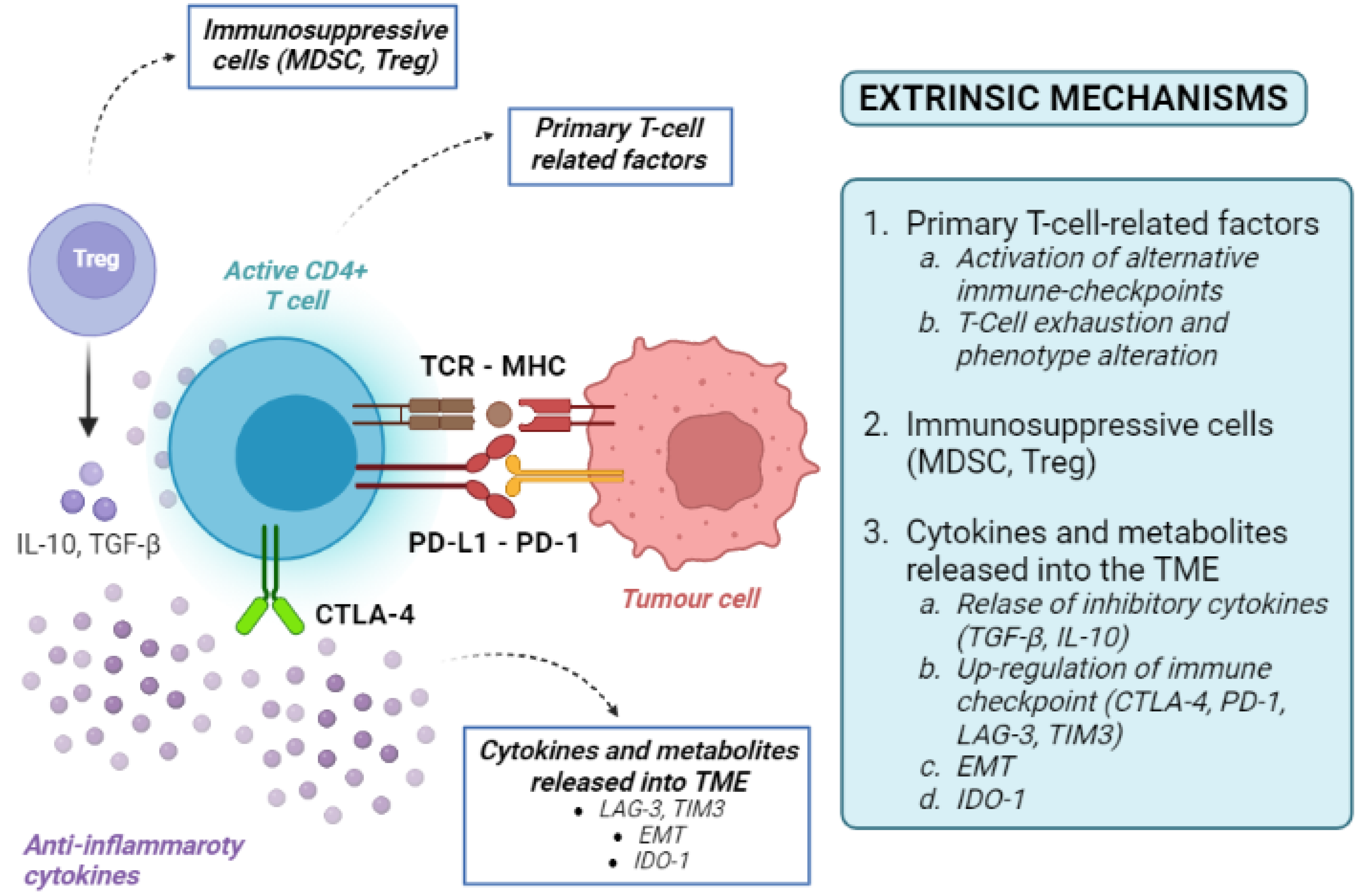
5. Mechanisms of Acquired ICI Resistance
Although the specific mechanisms of acquired (secondary) resistance are poorly understood, some of these are overlapped in primary and acquired ICI resistance. In the specific case of HCC, these mechanisms are still elusive. Tumour cells can lose some encoding sequences for tumour neo-antigens through the elimination of specific sub-clones or the deletion of chromosomal regions, resulting in the selection of less immunogenic tumour cell clones spared from T-cell killing [54][55][54,55]. For example, an acquired homozygous loss of beta-2 microglobulin, decreasing the expression of HLA and disrupting the antigen presentation, was identified in lung cancer. Defective HLA class I antigen processing through deleterious mutations in beta-2 microglobulin has been shown in melanoma [56][57][56,57]. Regarding melanoma, acquired resistance to PD-1 inhibitors can also be mediated by JAK1/2-inactivating mutations. The up-regulation of both immune checkpoints and immune-suppressive cytokines may be other causes of acquired resistance. For instance, lung cancer patients with acquired resistance against anti-PD1 therapy show an up-regulation of PD-1 [9]. Although it is still unclear whether PD-L1 expression causes an increased or decreased ICI response, it could result in the suppressed antitumour effect of T cells [58].6. Current Strategies in Immunotherapy for HCC
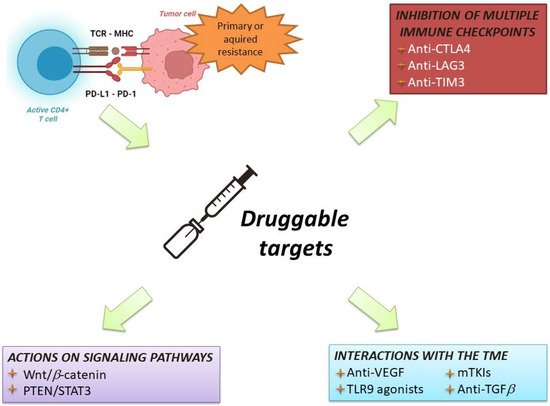
The initial studies of immunotherapy in HCC patients included ICI monotherapies [59]. Despite promising results of Phase 2 trials, two different Phase 3 randomised clinical trials failed to show the superiority of nivolumab vs. sorafenib in the frontline setting, and pembrolizumab vs. placebo in the second line setting, respectively [60][61][60,61]. Nevertheless, trials of ICI monotherapies provided three pieces of important information: First, these regimens were safe and well-tolerated, even in patients with liver cirrhosis and chronic viral infections. Second, ICIs obtained an objective response rate in 15–18% of cases [60][61][60,61], which is considerably higher that of TKI. Third, patients with an objective response had very long survival times [62]. Therefore, associating ICIs with agents able to overcome resistance to immunotherapy has become the most common strategy to improve the response rate and survival of HCC patients (Figure 4).