 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jae Chan Chang | -- | 4446 | 2022-10-11 04:04:14 | | | |
| 2 | Conner Chen | + 1 word(s) | 4447 | 2022-10-11 10:24:10 | | |
Video Upload Options
Endotheliopathy, according to the “two-activation theory of the endothelium”, is triggered by the activated complement system in critical illnesses, such as sepsis, diabetes and polytrauma, leading to two distinctly different molecular dysfunctions: (1) the activation of the inflammatory pathway due to the release of inflammatory cytokines, such as interleukins, interferons and tumor necrosis factors, and (2) the activation of the microthrombotic pathway due to the exocytosis of hemostatic factors, including ultra-large von Willebrand factor (ULVWF) multimers and FVIII. These lead to inflammation and microthrombogenesis. The former produces inflammatory diseases, and the latter produces endotheliopathy-associated vascular microthrombotic disease (EA-VMTD), which orchestrates not only TTP-like syndrome characterized by the triad of consumptive thrombocytopenia, microangiopathic hemolytic anemia and multiorgan dysfunction syndrome, but also many other endotheliopathic syndromes. The diagnostic features of EA-VMTD are well established now and therapeutic strategies are being formulated.
1. Introduction
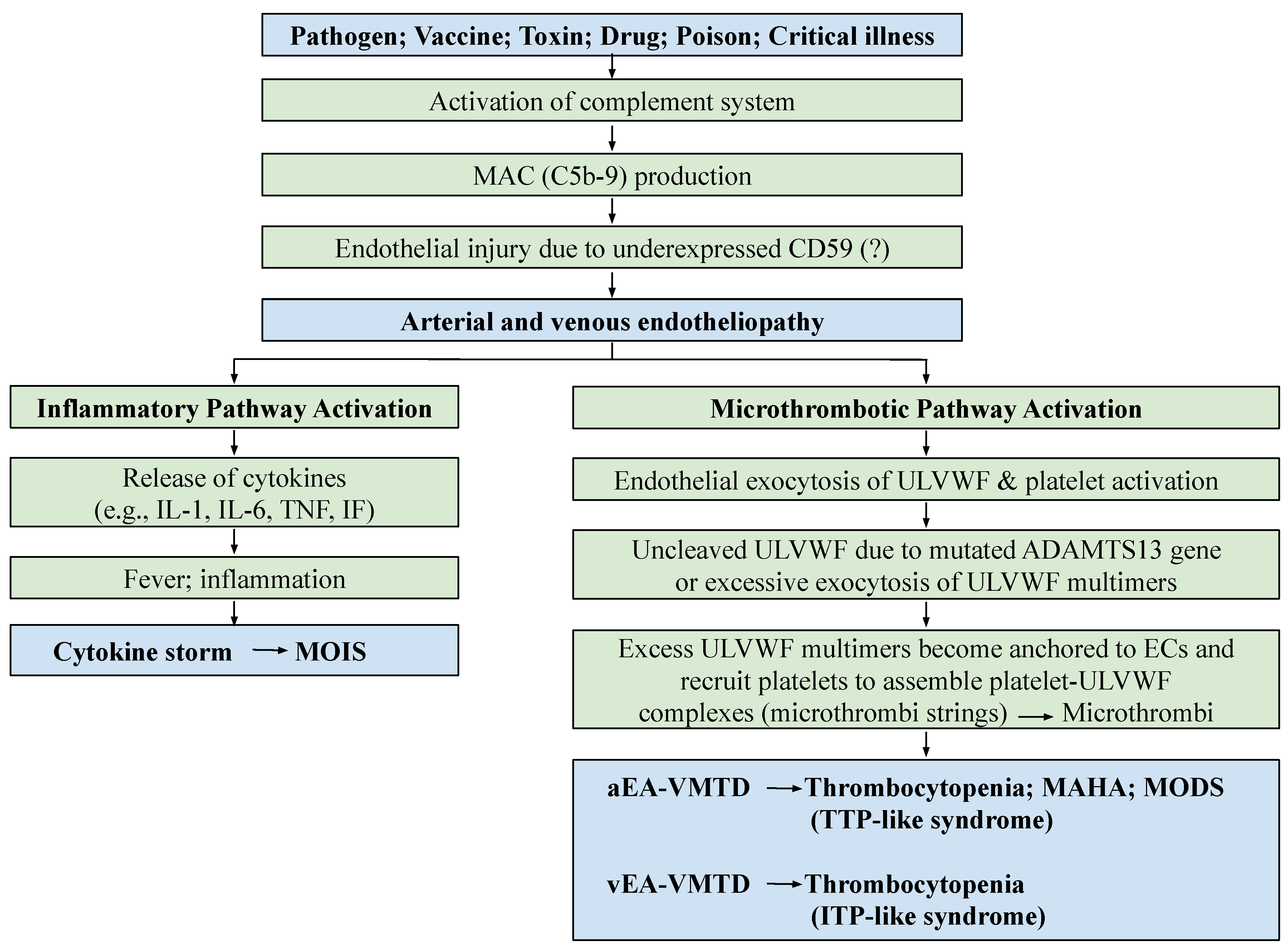
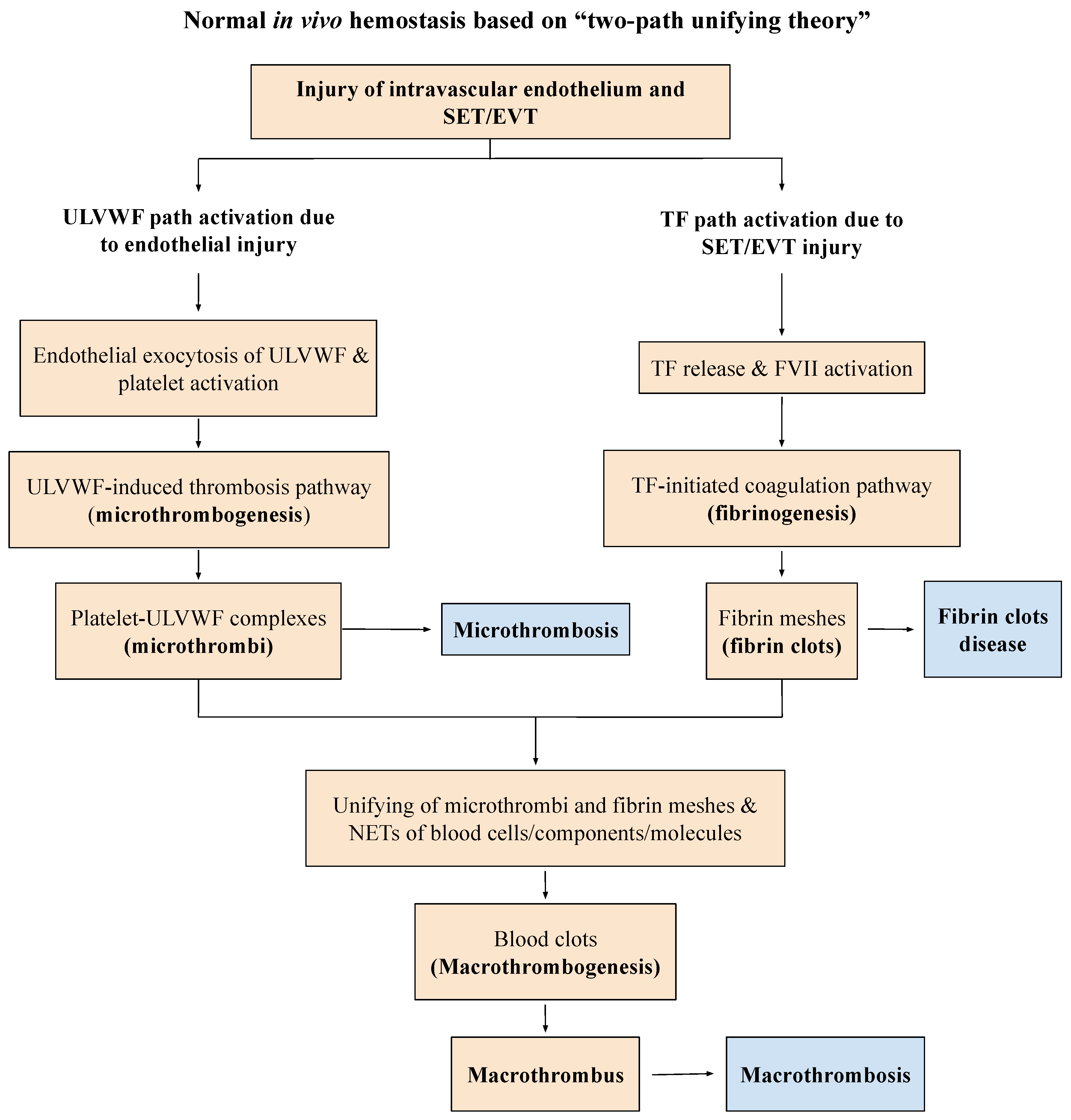
2. Genesis of Endotheliopathy
2.1. Causes of Endotheliopathy
2.1.1. Complement System
2.1.2. Endothelial Molecular Pathogenesis
| Hemostasis | Coagulation | Thrombosis | |
|---|---|---|---|
| Term concept | Philosophical | Physiological | Structural |
| Implied meaning | Natural process in vivo | Artificial process in vitro Physiologic process at bleeding site |
Pathological process in vivo |
| Involved site | Blood-vessel wall | Test tube Extravascular trauma to vessel wall |
Intravascular lumen |
| Products | Hemostatic plug | Fibrin mesh/fibrin clot * | Microthrombi/macrothrombi |
| Critical role | Vascular-wall injury | Coagulation test Hemorrhage |
Physiologic thrombogenesis |
| Components | Endothelium SET EVT Blood in circulation |
TF/thromboplastin Coagulation proteins/serine proteases |
ULVWF/FVIII from ECs Platelets+ from circulation Coagulation proteins/serine proteases TF from SET/EVT |
| Phenotypes | Determined by the level of vascular damage |
Determined by participating coagulation factors |
Determined by ULVWF, platelets, TF, hemostatic factors, and unifying mechanism |
| Inciting example | Endotheliopathy Vascular injury |
Coagulation tests for PT and aPTT | VMTD/MODS due to microthrombosis EA-VMTD: TTP-like syndrome: “DIC” Arterial macrothrombosis VTE Combined micro–macrothrombosis |
2.2. Genesis of Clinical Phenotypes in Endotheliopathy
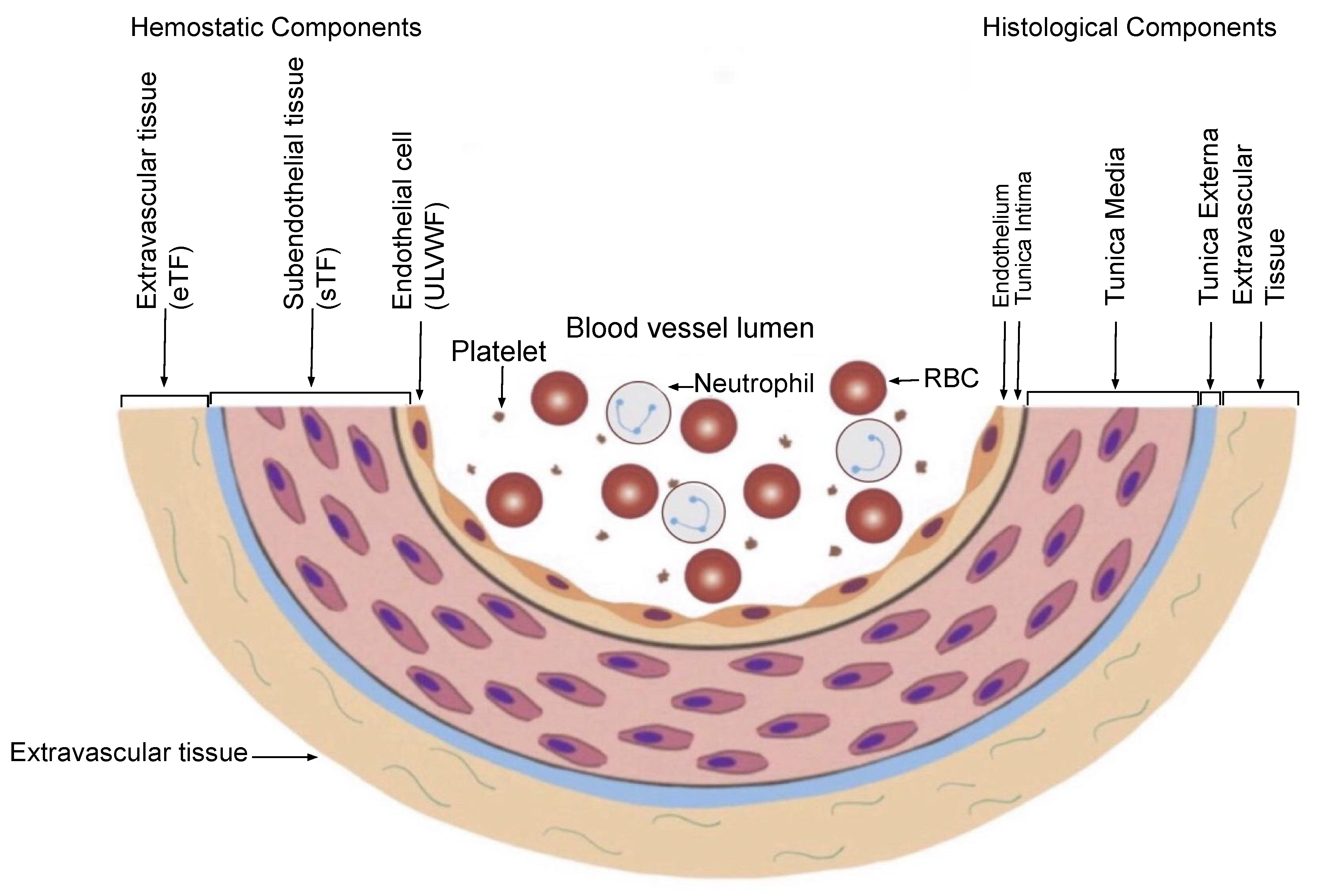
2.2.1. Hemostasis Based on the Blood-Vessel Model
| (1) Hemostatic Principles | ||
|
||
| (2) Major Participating Components | ||
| Components | Origin | Mechanism |
| (1) ECs/SET/EVT | Blood-vessel wall/EVT | Protective barrier |
| (2) ULVWF | ECs | Endothelial exocytosis/anchoring and microthrombogenesis |
| (3) Platelets | Circulation | Adhesion to ULVWF strings/assembling and microthrombogenesis |
| (4) TF | SET and EVT | Release from tissue due to vascular injury/leading and fibrinogenesis |
| (5) Coagulation factors | Circulation | Activation of coagulation factors/forming and macrothrombogenesis |
| (3) Vascular Injury and Hemostatic Phenotypes | ||
| Injury-Induced Damage | Involved Hemostatic Path | Level of Vascular Injury and Examples |
| (1) Endothelium | ULVWF | Level 1 damage—microthrombosis (e.g., TIA [focal]; Heyde’s syndrome [local); EA-VMTD [disseminated]) |
| (2) Endothelium/SET | ULVWF + sTF | Level 2 damage—macrothrombosis (e.g., AIS; DVT; PTE; AA) |
| (3) Endothelium/SET/EVT | ULVWF + eTF | Level 3 damage—macrothrombosis with hemorrhage (e.g., THS; THMI) |
| (4) EVT alone | eTF | Level e damage—fibrin clot disease (e.g., AHS (e.g., SDH; EDH); ICH; organ/tissue hematoma) |
| Hemostatic Phenotypes | Causes | Genesis |
| (1) Hemorrhage | External bodily injury | Trauma-induced external bleeding (e.g., accident; assault; self-inflicted injury) |
| (2) Hematoma | Internal EVT injury | Obtuse trauma-induced bleeding (e.g., tissue and cavitary hematoma; hemarthrosis) |
| (3) Thrombosis | Intravascular injury | Intravascular injury (e.g., atherosclerosis; diabetes; indwelling venous catheter; surgery; vascular access) |
2.2.2. Vascular Injury Provoking Thrombosis
- Microthrombi strings composed of platelet–ULVWF complexes [32][33];
- Documented endothelial exocytosis of ULVWF and FVIII in patient with sepsis and other endotheliopathy [34][35][36];
- The well-known role of ULVWF from ECs in the early phase of hemostasis in external bodily injury;
- Prominent interaction between platelets and ULVWF in a vascular injury, resulting in consumptive thrombocytopenia [37][38];
- The lack of participation of TF in hemostasis producing septic microthrombosis [4];
- TF unavailable in the EC injury but available from the SET/EVT in local bleeding vascular injury;
- Distinctly different disseminated microthrombosis occurring in the microvasculature in sepsis compared with localized macrothrombosis formed after fibrin clots occurring in a local large-vessel injury;
- The irrefutable hemostatic fundamental: “The hemostasis and thrombogenesis can be activated only by a vascular injury”.
- Damaged ECs from the blood-vessel wall following a vascular injury release ULVWF/FVIII in partnership, which are the essential components, along with platelets, in the formation of microthrombi strings. The process of ULVWF interacting with platelets is called “microthrombogenesis”;
- Damaged SET from the blood-vessel wall following a local vascular injury releases TF into circulation, which activates FVIIa and leads to the formation of fibrin clots via the extrinsic cascade with the sequential activation of FIX, FX, FV, FII, and fibrinogen to fibrin. The process is called “fibrinogenesis”;
- A local traumatic vascular injury involving ECs and SET produces both microthrombi strings and fibrin clots. The proposed theory is that the forming process of macrothrombosis must be the result of the “unifying mechanism of microthrombi strings and fibrin meshes”. This process is called “macrothrombogenesis”, in which neutrophil extracellular traps (NETosis) passively participate;
- The understanding of this unifying mechanism is very important in the understanding of arterial and venous combined micro–macrothrombosis, which is a process that should be called micro–macrothrombogenesis.
| Thrombosis Mechanism | Microthrombogenesis | Fibrinogenesis | Macrothrombogenesis |
|---|---|---|---|
| Utilizing hemostatic path | ULVWF path | TF path | Combined ULVWF and TF path |
| Examples | Sepsis (bacterial, viral, fungal, rickettsial, parasitic) Polytrauma, surgery, transplant Toxin, drug, venom, vaccine Pregnancy Diseases (autoimmune, cancer, diabetes) |
APL SDH EDH |
DVT (distal) VTE (central DVT/PTE) Arterial thrombosis Peripheral gangrene |
| Thrombosis character | Microthrombi strings | Fibrin clots | Macrothrombus Combined micro–macrothrombi |
| Vascular lesion and phenotype example | Focal—retinal microaneurysm Local—hepatic VOD Multifocal—HERNS, Susac syndrome Disseminated—EA-VMTD/MODS |
Disseminated—true DIC | Local—distal DVT, arterial thrombosis, AIS |
| Complex vascular/ hematologic phenotypes |
Venous—venous microthrombi (ITP-like syndrome) Arterial—arterial microthrombi (TTP-like syndrome, MODS) |
Venous/arterial—DIC (fibrin clot disease) | Combined micro–macrothrombosis Venous—VTE, PTE, CVST Arterial—SPG, limb gangrene |
References
- Hattori, R.; Hamilton, K.K.; McEver, R.P.; Sims, P.J. Complement proteins C5b-9 induce secretion of high molecular weight mul-timers of endothelial von Willebrand factor and translocation of granule membrane protein GMP-140 to the cell surface. J. Biol. Chem. 1989, 264, 9053–9060.
- Brunn, G.J.; Saadi, S.; Platt, J.L. Differential Regulation of Endothelial Cell Activation by Complement and Interleukin 1α. Circ. Res. 2006, 98, 793–800.
- Kerr, H.; Richards, A. Complement-mediated injury and protection of endothelium: Lessons from atypical haemolytic uraemic syndrome. Immunobiology 2012, 217, 195–203.
- Chang, J.C. Thrombocytopenia in critically ill patients due to vascular microthrombotic disease: Pathogenesis based on “two activation theory of the endothelium”. Vasc. Dis. Ther. 2017, 2, 2–7. Available online: https://www.oatext.com/pdf/VDT-2-132.pdf (accessed on 13 September 2022).
- Chang, J.C. TTP-like syndrome: Novel concept and molecular pathogenesis of endotheliopathy-associated vascular micro-thrombotic disease. Thromb. J. 2018, 16, 20.
- Chang, J.C. Sepsis and septic shock: Endothelial molecular pathogenesis associated with vascular microthrombotic disease. Thromb. J. 2019, 17, 10.
- Chang, J.C. Acute Respiratory Distress Syndrome as an Organ Phenotype of Vascular Microthrombotic Disease: Based on Hemostatic Theory and Endothelial Molecular Pathogenesis. Clin. Appl. Thromb. Hemost. 2019, 25, 1076029619887437.
- Chang, J.C. Disseminated intravascular coagulation: New identity as endotheliopathy-associated vascular microthrombotic disease based on in vivo hemostasis and endothelial molecular pathogenesis. Thromb. J. 2020, 18, 25.
- Chang, J. Pathogenesis of Ebola Viral Haemorrhagic Fever: TTP-like Syndrome Associated with Hepatic Coagulopathy based on “Two Activation Theory of the Endothelium”. J. Prev. Infect. Control 2017, 3, 1–4.
- Chang, J.C. COVID-19 Sepsis: Pathogenesis and Endothelial Molecular Mechanisms Based on “Two-Path Unifying Theory” of Hemostasis and Endotheliopathy-Associated Vascular Microthrombotic Disease, and Proposed Therapeutic Approach with Antimicrothrombotic Therapy. Vasc. Health Risk Manag. 2021, 17, 273–298.
- Chang, J.C. Hemostasis based on a novel ‘two-path unifying theory’ and classification of hemostatic disorders. Blood Coagul. Fibrinolysis 2018, 29, 573–584.
- Chang, J.C. Thrombogenesis and thrombotic disorders based on “two-path unifying theory of hemostasis: Philosophical, physiological and phenotypical interpretation. Blood Coagul. Fibrinolysis 2018, 29, 585–595.
- Chang, J.C.; Hawley, H.B. Vaccine-Associated Thrombocytopenia and Thrombosis: Venous Endotheliopathy Leading to Venous Combined Micro-Macrothrombosis. Medicina 2021, 57, 1163.
- Chang, J.C. Pathogenesis of Two Faces of DVT: New Identity of Venous Thromboembolism as Combined Micro-Macrothrombosis via Unifying Mechanism Based on “Two-Path Unifying Theory” of Hemostasis and “Two-Activation Theory of the Endothelium”. Life 2022, 12, 220.
- Esmon, C.T. Crosstalk between inflammation and thrombosis. Maturitas 2004, 47, 305–314.
- Aksu, K.; Donmez, A.; Keser, G. Inflammation-induced thrombosis: Mechanisms, disease associations and management. Curr. Pharm. Des. 2012, 18, 1478–1493.
- Engelmann, B.; Massberg, S. Thrombosis as an intravascular effector of innate immunity. Nat. Rev. Immunol. 2013, 13, 34–45.
- Allam, R.; Kumar, S.V.; Darisipudi, M.N.; Anders, H.-J. Extracellular histones in tissue injury and inflammation. J. Mol. Med. 2014, 92, 465–472.
- Palankar, R.; Greinacher, A. Challenging the concept of immunothrombosis. Blood 2019, 133, 508–509.
- Chang, J.C. Disseminated intravascular coagulation (DIC): Is it fact or fancy? Blood Coagul. Fibrinolysis 2018, 29, 330–337.
- Turner, N.A.; Moake, J. Assembly and activation of alternative complement components on endothelial cell-anchored ultra-large von Willebrand factor links complement and hemostasis-thrombosis. PLoS ONE 2013, 8, e59372.
- Amari Chinchilla, K.; Vijayan, M.; Taveras Garcia, B.; Jim, B. Complement-Mediated Disorders in Pregnancy. Adv. Chronic Kidney Dis. 2020, 27, 155–164.
- Ghosh, P.; Sahoo, R.; Vaidya, A.; Chorev, M.; Halperin, J.A. Role of Complement and Complement Regulatory Proteins in the Complications of Diabetes. Endocr. Rev. 2015, 36, 272–288.
- Xie, C.B.; Jane-Wit, D.; Pober, J.S. Complement Membrane Attack Complex: New Roles, Mechanisms of Action, and Therapeutic Targets. Am. J. Pathol. 2020, 190, 1138–1150.
- Yu, J.; Abagyan, R.; Dong, S.; Gilbert, A.; Nussenzweig, V.; Tomlinson, S. Mapping the Active Site of CD59. J. Exp. Med. 1997, 185, 745–754.
- Wu, G.; Hu, W.; Shahsafaei, A.; Song, W.; Dobarro, M.; Sukhova, G.K.; Bronson, R.R.; Shi, G.-P.; Rother, R.P.; Halperin, J.A.; et al. Complement Regulator CD59 Protects Against Atherosclerosis by Restricting the Formation of Complement Membrane Attack Complex. Circ. Res. 2009, 104, 550–558.
- Blann, A.D. How a damaged blood vessel wall contibutes to thrombosis and hypertension. Pathophysiol. Haemost. Thromb. 2003, 33, 445–448.
- Teijaro, J.R.; Walsh, K.B.; Cahalan, S.; Fremgen, D.M.; Roberts, E.; Scott, F.; Martinborough, E.; Peach, R.; Oldstone, M.B.; Rosen, H. Endothelial cells are central orchestrators of cytokine amplification during influenza virus infection. Cell 2011, 146, 980–991.
- Adam, E.; Zacharowski, K.; Miesbach, W. A comprehensive assessment of the coagulation profile in critically ill COVID-19 patients. Thromb. Res. 2020, 194, 42–44.
- Favaloro, E.J.; Henry, B.M.; Lippi, G. Increased VWF and Decreased ADAMTS-13 in COVID-19: Creating a Milieu for (Micro) Thrombosis. Semin. Thromb. Hemost. 2021, 47, 400–418.
- Bagot, C.N.; Arya, R. Virchow and his triad: A question of attribution. Br. J. Haematol. 2008, 143, 180–190.
- Chauhan, A.K.; Goerge, T.; Schneider, S.W.; Wagner, D.D. Formation of platelet strings and microthrombi in the presence of ADAMTS-13 inhibitor does not require P-selectin or beta3 integrin. J. Thromb. Haemost. 2007, 5, 583–589.
- Tsai, H.-M. Pathophysiology of thrombotic thrombocytopenic purpura. Int. J. Hematol. 2010, 91, 1–19.
- Nguyen, T.C.; Liu, A.; Liu, L.; Ball, C.; Choi, H.; May, W.S.; Aboulfatova, K.; Bergeron, A.L.; Dong, J.-F. Acquired ADAMTS-13 deficiency in pediatric patients with severe sepsis. Haematologica 2007, 92, 121–124.
- Karim, F.; Adil, S.N.; Afaq, B.; Haq, A.U. Deficiency of ADAMTS-13 in pediatric patients with severe sepsis and impact on in-hospital mortality. BMC Pediatr. 2013, 13, 44.
- Fukushima, H.; Nishio, K.; Asai, H.; Watanabe, T.; Seki, T.; Matsui, H.; Sugimoto, M.; Matsumoto, M.; Fujimura, Y.; Okuchi, K. Ratio of von Willebrand Factor Propeptide to ADAMTS13 Is Associated with Severity of Sepsis. Shock 2013, 39, 409–414.
- Mendolicchio, G.L.; Ruggeri, Z.M. Interaction of von Willebrand factor with platelets and the vessel wall. Hämostaseologie 2015, 35, 211–224.
- Shim, C.Y.; Ni Liu, Y.; Atkinson, T.; Xie, A.; Foster, T.; Davidson, B.P.; Treible, M.; Qi, Y.; López, J.A.; Munday, A.; et al. Molecular Imaging of Platelet–Endothelial Interactions and Endothelial von Willebrand Factor in Early and Mid-Stage Atherosclerosis. Circ. Cardiovasc. Imaging 2015, 8, e002765.
- Stein, E.; McMahon, B.; Kwaan, H.; Altman, J.K.; Frankfurt, O.; Tallman, M.S. The coagulopathy of acute promyelocytic leukaemia revisited. Best Pract. Res. Clin. Haematol. 2009, 22, 153–163.
- Chang, J.C. Stroke Classification: Critical Role of Unusually Large von Willebrand Factor Multimers and Tissue Factor on Clinical Phenotypes Based on Novel “Two-Path Unifying Theory” of Hemostasis. Clin. Appl. Thromb. Hemost. 2020, 26, 1076029620913634.

