2. Genesis of Endotheliopathy
The endothelium is a unique single layer of cells lining between the blood in circulation and the subendothelial tissue of every organ to protect the body from the intrusion of harmful external toxic molecules and microorganisms. It also provides the direct and indirect contact of nutrients and metabolic components to the subendothelial and extravascular tissues to sustain life, and it is the barrier to and partner with the blood cells to maintain hemostasis and physiologic homeostasis. Therefore, an insult causing endothelial damage disrupts the normal anatomy and physiologic function. The endothelium may lead to diversified paths of the pathogenesis that trigger the dysfunction of the cells, tissues, organs, and multisystem. Until now, clinical medicine has designated most human diseases with terms that define and appropriate the dysfunction of the cell (e.g., microangiopathic hemolytic anemia), tissue (e.g., rhabdomyolysis), organ (e.g., acute kidney failure), and multisystem (e.g., systemic lupus erythematosus). However, clinical medicine has not been able to put together the etiologic identities and pathogeneses of many clinical syndromes in relation to molecular, metabolic, and structural alteration. This paucity of
our knowledge in endotheliopathy has also entailed the utilization of imprecise terms for diagnosis, such as Kawasaki disease, acute respiratory distress syndrome (ARDS), multisystem inflammatory syndrome in child (MIS-C), thromboangiitis obliterans (e.g., Buerger’s disease), cerebral venous sinus thrombosis (CVST), DIC-like coagulopathy, limb gangrene, and others, to define the clinical syndromes that have been occurring during the COVID-19 pandemic and in other diseases
[10].
2.1. Causes of Endotheliopathy
Endotheliopathy is not a distinctive disease that delineates the dysfunction of a specific cell type, tissue, organ, or system, but an intermediary pathology that leads to almost every human disease involving the vascular-tree system. To date, the accepted medical tenet has been that endothelial damage results in a transient endothelial intravascular injury, typically due to localized vascular pathology, and often with limited clinical consequence. However, now it is abundantly clear that “endotheliopathy” could become a major pathology that produces life-threatening cellular, tissue, and organ damage, leading to their dysfunction, and even to multisystem disease
[4][5][6][7][8][9][10][4,5,6,7,8,9,10]. The endothelium is the most important structure protecting lives, in which the pathology of many human diseases could originate or be influenced. Its integrity often determines the clinical outcomes of critical illnesses.
2.1.1. Complement System
Endotheliopathy is triggered by the activated complement system in response to pathogens and other insults
[3][6][21][3,6,21]. Complement activation can occur through one of three different pathways (i.e., classical, alternate, and lectin). The complement system can not only be activated by an external insult, such as a pathogen, toxin or drug, vaccine, venom, surgery, transplant, or polytrauma, but also by internal physiologic changes, such as pregnancy, hypertension, hyperglycemia, as well as the autoimmune process
[5][6][22][23][5,6,22,23].
The lytic function of the complement membrane attack complex (MAC: C5b-9) serves as an important defense against bacteria, viruses, and parasites, and the genetic deficiency of the terminal complement proteins C5 through C9 predisposes the host to recurrent infections
[24]. The innate immune response of complement activation is not only the first step in protecting the host against a variety of pathogens, but it may also play a detrimental role in innocent bystander endothelial cells (ECs). Should the unchecked production of MAC be deposited to the endothelial membrane, channel formation could be formed and lead to endothelial dysfunction in the host
[3]. When MAC provokes either structural or functional endothelial damage in the host, it activates at least two unique molecular mechanisms, which promote clinically significant endotheliopathic syndromes
[4][5][6][7][4,5,6,7].
2.1.2. Endothelial Molecular Pathogenesis
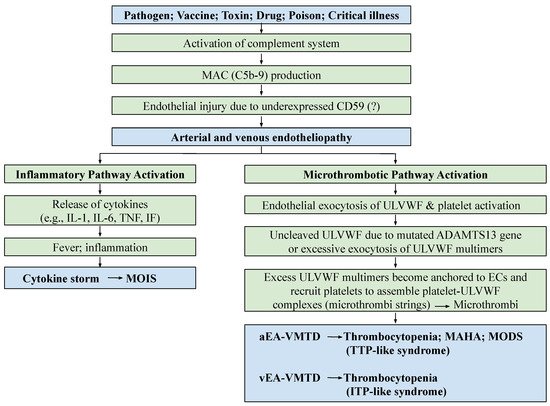
The molecular pathogenesis of endotheliopathy is elaborated in the “two-activation theory of the endothelium”, which shows complement-induced endothelial molecular events (
Figure 1), leading to the activation of: (1) the inflammatory pathway, and (2) the microthrombotic pathway
[4]. MAC-induced endotheliopathy is suspected to occur if the endothelium is “unprotected” due to downregulated CD59
[25][26][25,26].
Both functional and structural injuries of the endothelium release several mediators altering homeostasis and activate a host of biomolecules expressed in ECs, which can promote, directly and indirectly, the creation of different pathologic and clinical phenotypes. The numerous biomolecules that are released from ECs include inflammatory cytokines, hemostatic factors, and adhesive molecules, which are interleukins (IL), tumor necrosis factors (TNF), interferons (IFN), coagulation factors, tissue plasminogen activator, prostacyclin, nitric oxide, and others
[6][7][27][6,7,27]. Although their roles in vascular disease have not been well defined yet, the pathogenesis of inflammation is postulated to be due to released cytokines and chemokines, such as interleukins IL-1, IL-2, IL-6, TNF, IFN, and others, and microthrombogenesis is promoted by ULVWF multimers and hemostatic factors, resulting from endotheliopathy
[5][6][7][8][9][10][28][5,6,7,8,9,10,28]. These molecular pathogeneses were predicted before the onset of the COVID-19 pandemic
[6][7][6,7], and important molecular events have been unequivocally confirmed during the pandemic
[29][30][29,30].
The mechanism of molecular pathogenesis in disseminated arterial and venous endotheliopathy based on the “two-activation theory of the endothelium” is shown in
Figure 1. On the one hand, the activated inflammatory pathway provokes inflammation due to released inflammatory cytokines in sepsis, or other critical illnesses. On the other hand, the activated microthrombotic pathway triggers the excessive release of ULVWF/FVIII from endothelial exocytosis
[5][6][5,6]. Should a relative insufficiency of ADAMTS13 be present due to the heterozygous gene mutation or polymorphism of the gene, and/or due to the excess release of ULVWF multimers, partial hemostasis of lone ULVWF path becomes activated and ULVWF attract platelets, which leads to the formation of “microthrombi strings” composed of platelet–ULVWF complexes
[5][6][5,6]. This process is called microthrombogenesis, and leads to endotheliopathy-associated disseminated vascular microthrombotic disease (EA-VMTD)
[5][6][11][5,6,11].
This partial hemostasis due to lone activation of the ULVWF path is characterized by consumptive thrombocytopenia, overexpressed ULVWF/VWF/VWF antigen and increased activity of FVIII due to their endothelial release, resulting in relative ADAMTS13 insufficiency. Endotheliopathy is systemic in sepsis and disease due to certain drugs or toxins, and it is clinically disseminated in the microvasculature, promoting “microvascular microthrombosis” (i.e., EA-VMTD). In the past, the “coagulopathy” occurring in the septic patient was thought to be due to the overactivation of the tissue factor (TF) path leading to systemic “fibrin clots” formation, and had been called “DIC”. However, its pathogenesis has never been clearly established. Based on the two novel hemostatic theories, “DIC” has been identified as a form of thrombosis, which is exclusively due to “microthrombosis” caused by the activated ULVWF path that develops from the injury of ECs
[5][6][8][20][5,6,8,20]. The conflict in the terms between “coagulation” and “thrombosis” related to “hemostasis” has contributed to the misunderstanding of the pathogenesis of “DIC”, and of the identification of the pathophysiological mechanism of hemostasis in vivo. Thus, in the past, the true meaning and genesis of “fibrin clots” and “microthrombi” could not been conceptualized, which contributed to uncomfortable debates between coagulopathy and thrombosis (thrombopathy) in the coagulation community. To clarify the different meanings of their conceptual terms, I have briefly summarized the discriminating interpretations of the terms in
Table 1.
Table 1.
Conceptual understanding of the terms: hemostasis, coagulation, and thrombosis.
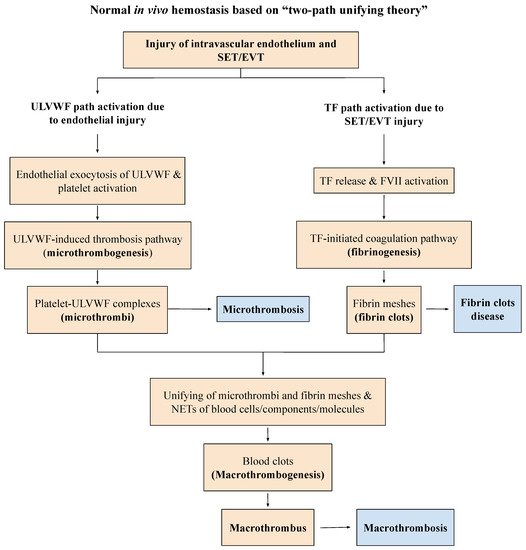
Many medical studies have emphasized that the increased activities of FVIII and VWF in critical illnesses are reactive changes in response to inflammation promoting the “hypercoagulable state” of “DIC”. This interpretation has led to a conceptually and structurally wrong conclusion on the nature of microthrombi because the increased expressions of FVIII and VWF are always the result of endothelial exocytosis from endotheliopathy, which is perfectly consistent with vascular injury limited to ECs producing “microthrombi strings”, as shown in the “two-path unifying theory” of hemostasis (
Figure 2)
[11][12][11,12].
2.2. Genesis of Clinical Phenotypes in Endotheliopathy
Endotheliopathy, even generalized, only activates lone ULVWF path of hemostasis because the complement-activated endothelial damage that releases biomolecules due to the pathogen toxin is limited to ECs, without affecting the subendothelial tissue (SET) and extravascular tissue (EVT)
[6]. Disseminated microthrombosis is made of microthrombi strings in the microvasculature because SET/EVT damage that releases TF to the intravascular space does not occur, and fibrin clots are not produced to form complete blood clots (i.e., macrothrombosis), as presented in
Figure 2.
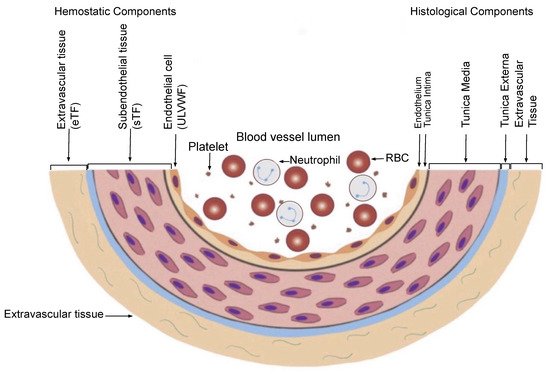
In contrast, a bleeding intravascular injury that causes both local EC and SET/EVT damage typically occurs in local trauma following local vascular damage. An in-hospital vascular access/device, or indwelling central vascular catheter, which releases not only a small amount of ULVWF/FVIII from local ECs, but also releases sufficient TF from SET and EVT, produces macrothrombosis. The relationship between ULVWF release from ECs and TF release from ECs and SET/EVT from their respective damage is illustrated in
Figure 3. In sepsis, endotheliopathy is disseminated even though its extent and phenotype expression are dependent upon endothelial heterogeneity and the tropism of the pathogen. When microthrombogenesis is triggered, disseminated microthrombosis causes clinical phenotypes by affecting different organs and producing organ dysfunction syndrome, including MODS. Furthermore, more complex phenotypes of microthrombosis can occur depending on: (1) the extent of the vascular-tree involvement (e.g., the multiplicity of the tissues and organs), (2) the localization of different vascular functional characters (e.g., arterial vs. venous system), and (3) an additional interacting mechanism with an underlying gene mutation or environmental factor (e.g., thrombophilia, such as FV Leiden, or protein C deficiency, and von Willebrand disease, pathogens, and toxins)
[13]. All of these different phenotypes that occur in endotheliopathy-associated “focal”, “multifocal”, “localized”, “regionalized”, and “disseminated” microthrombosis are inclusively called EA-VMTD
[5].
Figure 3. Schematic illustration of cross section of blood-vessel histology and hemostatic components (reproduced and modified with permission from Chang JC.
Clin Appl Thromb Hemost 2019 January–December; 25:1076029619887437). The blood-vessel wall is the site of hemostasis (coagulation) in the external bodily injury at which blood clots (hemostatic plug) are produced and hemorrhage is stopped. It is also the site of hemostasis (thrombogenesis) in the intravascular injury in which intravascular blood clots (thrombus) are produced to cause thrombosis. Its histologic components are divided into the endothelium, tunica intima, tunica media, and tunica externa, and each component has its function that contributes to molecular hemostasis. As illustrated, EC damage triggers the exocytosis of ULVWF and SET damage, and it promotes the release of sTF from the tunica intima, tunica media, and tunica externa. EVT damage releases eTF from the outside of the blood-vessel wall. This depth of the blood-vessel-wall injury contributes to the genesis of different thrombotic disorders, such as microthrombosis, macrothrombosis, fibrin clots, thrombo-hemorrhagic clots, and various endotheliopathic syndromes. This concept based on the blood-vessel-wall model is especially important in the understanding of different phenotypes of stroke and heart attack. Abbreviations: EVT: extravascular tissue; eTF: extravascular tissue factor; SET: subendothelial tissue; sTF: subendothelial tissue factor; RBC: red blood cells; ULVWF: ultra-large von Willebrand factor.
2.2.1. Hemostasis Based on the Blood-Vessel Model
The concept of EA-VMTD cannot be understood without the comprehension of the hemostatic mechanism in vivo. However, hemostasis based on the blood-vessel model is simple and logical if the anatomy and physiology of the blood-vessel wall are understood, as illustrated in
Figure 3, and the three fundamentals in hemostasis are applied to the thrombogenesis, as summarized in
Table 2. The first and second principles illustrate how the molecular event of hemostatic components is initiated in the vascular-wall injury, and the third principle elaborates the physiologic paths of hemostasis that produces the pathologic phenotypes following an intravascular injury
[7].
Table 2.
Three fundamentals in normal hemostasis.
2.2.2. Vascular Injury Provoking Thrombosis
The established dogma of Virchow’s triad on thrombosis and thrombogenesis, which includes: (1) endothelial injury, (2) the stasis of blood flow, and (3) hypercoagulability, is considered the essential three components initiating the formation of a thrombus
[31]. Unfortunately, this conceptual misunderstanding of hemostasis, thrombosis, and coagulation has not only confused the concepts and uses of the terms, “coagulation” and “thrombosis”, but it has also hampered the discovery process of the true hemostatic mechanism in vivo to date. This author has attempted to redefine the conceptual connotation of these terms, as shown in
Table 1.
After the reinterpretation of “DIC” utilizing the differentiating concepts between “coagulation” and “thrombosis”, and their implied meanings in relation to hemostasis in vivo and coagulation in vitro, the true identity of “DIC” was found to be “endotheliopathic microthrombosis” rather than the “uncontrolled” activation of coagulation in vitro or in vivo
[8][20][8,20]. In other words, “DIC” is an endothelial phenomenon caused by well-defined partial hemostatic process (i.e., ULVWF path) rather than vague in vivo coagulation disorder reflecting in vitro coagulation abnormalities. Thus, “DIC” has been newly defined as EA-VMTD, which explains not only microvascular microthrombosis (i.e., VMTD), but also confirms the molecular changes associated with the disease (i.e., thrombocytopenia, increased activity of FVIII, and overexpression of ULVWF/VWF antigen)
[5][6][8][20][5,6,8,20]. Furthermore, this author was able to link EA-VMTD (microthrombosis of endotheliopathy) to “DIC” (microthrombosis of sepsis) and could separate the “fibrin clots” of fibrin clot disease (i.e., acute promyelocytic leukemia) from the “microthrombi strings” of “DIC”, constructing two hemostatic paths in vivo based on the vascular-wall model after analyzing the following inexplicable hemostatic facts. Finally, identified are the true characters of “microthrombosis” and “fibrin clots” according to the “two-path unifying theory” of hemostasis
[11][12][11,12].
-
Damaged ECs from the blood-vessel wall following a vascular injury release ULVWF/FVIII in partnership, which are the essential components, along with platelets, in the formation of microthrombi strings. The process of ULVWF interacting with platelets is called “microthrombogenesis”;
- Microthrombi strings composed of platelet–ULVWF complexes
- Distinctly different disseminated microthrombosis occurring in the microvasculature in sepsis compared with localized macrothrombosis formed after fibrin clots occurring in a local large-vessel injury;
- The irrefutable hemostatic fundamental: “The hemostasis and thrombogenesis can be activated only by a vascular injury”.
-
Microthrombi strings composed of platelet–ULVWF complexes [32,33];
- Documented endothelial exocytosis of ULVWF and FVIII in patient with sepsis and other endotheliopathy
- [34]
-
Documented endothelial exocytosis of ULVWF and FVIII in patient with sepsis and other endotheliopathy [34,];
- 35
- ][36]
- The well-known role of ULVWF from ECs in the early phase of hemostasis in external bodily injury;
- Prominent interaction between platelets and ULVWF in a vascular injury, resulting in consumptive thrombocytopenia
- [
- 37
- The lack of participation of TF in hemostasis producing septic microthrombosis
- [
- 4
- ]
- ;
- TF unavailable in the EC injury but available from the SET/EVT in local bleeding vascular injury;
-
TF unavailable in the EC injury but available from the SET/EVT in local bleeding vascular injury;
-
-
Distinctly different disseminated microthrombosis occurring in the microvasculature in sepsis compared with localized macrothrombosis formed after fibrin clots occurring in a local large-vessel injury;
-
The irrefutable hemostatic fundamental: “The hemostasis and thrombogenesis can be activated only by a vascular injury”.
The three fundamentals in normal hemostasis are perfectly congruous with the “two-path unifying theory”
[7]. Further, the above factual components demonstrate that it is impossible to correctly redefine both “microthrombosis” and “macrothrombosis” using the contemporary hemostatic theory which is based on sequential activation via the extrinsic coagulation cascade initiated by the TF–FVIIa complex alone,
[11], as well as fibrin clot disease of acute promyelocytic leukemia (APL)
[39]. The “two-path unifying theory” of hemostasis can explain every hemostatic phenotype that occurs in microthrombosis, macrothrombosis, and the unique fibrin clot disease. A true in vivo hemostatic theory should be inclusive of the following molecular and physiologic changes of thrombogenesis
[6][10][13][14][6,10,13,14]:
- Damaged ECs from the blood-vessel wall following a vascular injury release ULVWF/FVIII in partnership, which are the essential components, along with platelets, in the formation of microthrombi strings. The process of ULVWF interacting with platelets is called “microthrombogenesis”;
- Damaged SET from the blood-vessel wall following a local vascular injury releases TF into circulation, which activates FVIIa and leads to the formation of fibrin clots via the extrinsic cascade with the sequential activation of FIX, FX, FV, FII, and fibrinogen to fibrin. The process is called “fibrinogenesis”;
-
Damaged SET from the blood-vessel wall following a local vascular injury releases TF into circulation, which activates FVIIa and leads to the formation of fibrin clots via the extrinsic cascade with the sequential activation of FIX, FX, FV, FII, and fibrinogen to fibrin. The process is called “fibrinogenesis”;
- A local traumatic vascular injury involving ECs and SET produces both microthrombi strings and fibrin clots. The proposed theory is that the forming process of macrothrombosis must be the result of the “unifying mechanism of microthrombi strings and fibrin meshes”. This process is called “macrothrombogenesis”, in which neutrophil extracellular traps (NETosis) passively participate;
-
-
A local traumatic vascular injury involving ECs and SET produces both microthrombi strings and fibrin clots. The proposed theory is that the forming process of macrothrombosis must be the result of the “unifying mechanism of microthrombi strings and fibrin meshes”. This process is called “macrothrombogenesis”, in which neutrophil extracellular traps (NETosis) passively participate;
- The understanding of this unifying mechanism is very important in the understanding of arterial and venous combined micro–macrothrombosis, which is a process that should be called micro–macrothrombogenesis.
The above three basic physiological mechanisms of hemostasis in vivo, constructed in
Figure 2 and
Table 3, can not only explain every hemorrhagic disease and thrombotic disorder, but it can help also interpreting additional complex hemostatic phenotypes in stroke, heart attack, venous thromboembolism (VTE), CVST, gangrene syndromes, and other tissue/organ-specific syndromes
[10][40][10,40]. Because EC damage is disseminated without SET damage in sepsis, but EC and SET damages are deeper but limited to localized vascular trauma, the microthrombi strings causing microthrombosis must be the result of microthrombogenesis triggered by endotheliopathy, but macrothrombus unified of microthrombi strings and fibrin clots is the result of fibrinogenesis and macrothrombogenesis in locality (
Table 3). Two hemostatic theories have been identified: (1) the “two-activation theory of the endothelium” to explain inflammation and microthrombosis (e.g., VMTD), and (2) the “two-path unifying theory” of hemostasis to explain the geneses of microthrombosis, fibrin clot disease, and macrothrombosis (e.g., DVT). The former is illustrated in
Figure 1, and the latter in
Figure 2. These two hemostatic theories are being applied in identifying the activated hemostatic mechanisms in the different phenotypes of hemostatic diseases. These mechanisms have correctly established the pathogeneses of many poorly defined thrombotic diseases, which include microthrombosis (e.g., septic endotheliopathy, ARDS, HUS, veno-occlusive disease (VOD)
[4][5][6][7][8][9][10][4,5,6,7,8,9,10], macrothrombosis (e.g., acute ischemic stroke, deep venous thrombosis)
[10][12][40][10,12,40], arterial combined micro–macrothrombosis (e.g., symmetrical peripheral gangrene)
[6][10][6,10], venous combined micro–macrothrombosis (e.g., VTE, CVST)
[13][14][13,14], MODS
[6], and ARDS, with complex forms of thrombosis during the COVID-19 pandemic
[7][10][7,10], including the following endotheliopathic syndromes in other human diseases.
Table 3. Three thrombogenetic paths and their clinical phenotypes of thrombosis according to the “two-path unifying theory of hemostasis”.