Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Brandon Lucke-Wold | -- | 1415 | 2022-10-11 03:20:06 | | | |
| 2 | Conner Chen | Meta information modification | 1415 | 2022-10-11 08:57:03 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Dodd, W.S.; Panther, E.J.; Pierre, K.; Hernandez, J.S.; Patel, D.; Lucke-Wold, B. Traumatic Brain Injury and Secondary Neurodegenerative Disease. Encyclopedia. Available online: https://encyclopedia.pub/entry/28764 (accessed on 10 August 2026).
Dodd WS, Panther EJ, Pierre K, Hernandez JS, Patel D, Lucke-Wold B. Traumatic Brain Injury and Secondary Neurodegenerative Disease. Encyclopedia. Available at: https://encyclopedia.pub/entry/28764. Accessed August 10, 2026.
Dodd, William S., Eric J. Panther, Kevin Pierre, Jairo S. Hernandez, Devan Patel, Brandon Lucke-Wold. "Traumatic Brain Injury and Secondary Neurodegenerative Disease" Encyclopedia, https://encyclopedia.pub/entry/28764 (accessed August 10, 2026).
Dodd, W.S., Panther, E.J., Pierre, K., Hernandez, J.S., Patel, D., & Lucke-Wold, B. (2022, October 11). Traumatic Brain Injury and Secondary Neurodegenerative Disease. In Encyclopedia. https://encyclopedia.pub/entry/28764
Dodd, William S., et al. "Traumatic Brain Injury and Secondary Neurodegenerative Disease." Encyclopedia. Web. 11 October, 2022.
Copy Citation
Traumatic brain injury (TBI) is a devastating event with severe long-term complications. TBI and its sequelae are one of the leading causes of death and disability in those under 50 years old. It is clear that neurotrauma can incite chronic neurodegenerative processes. Chronic traumatic encephalopathy, Parkinson’s disease, and many other neurodegenerative syndromes have all been associated with a history of traumatic brain injury.
neurotrauma
secondary mechanisms
neurodegeneration
1. Introduction
Neurodegeneration following traumatic brain injury is a complex process that is initiated by several distinct pathways which overwhelm homeostatic stress responses and trigger cellular degeneration and death. Recent studies have demonstrated a progression of neurodegenerative processes months and even years after traumatic brain injury, termed secondary neurodegeneration. Secondary neurodegeneration can manifest in many ways depending on specific etiology and affected neuroanatomy. Chronic traumatic encephalopathy (CTE) is a well-known disease closely associated with repeated traumatic brain injuries (TBI) [1][2]. Parkinson’s disease (PD), frontotemporal dementia (FTD), and other neurodegenerative diseases are less common but can also be induced as a consequence of TBI [3][4][5]. The precise incidence of CTE is hard to quantify due to diagnostic limitations; however, it has gained notoriety due to the prominence of repeated mild TBI in professional sports [1][6]. On the other hand, severe TBI with greater than 1 h loss of consciousness triples the risk of eventually developing PD [3]. Investigation into neurodegenerative disease secondary to TBI is rapidly evolving due to its complex pathophysiology and important public health implications.
2. Mechanisms of Neurodegeneration after TBI
The inciting mechanisms of secondary neurodegeneration after TBI are an interdependent set of pathological changes initiated by the primary traumatic injury (Figure 1). The temporal evolution of brain injury after TBI is multidimensional and complex but can be conceptualized as overlapping phases. The “acute injury” phase after TBI is characterized by the predominance of mechanical damage resulting from the initial trauma while the “secondary injury” phase is characterized by the delayed emergence of dysregulated metabolism and inflammation pathways [7][8][9]. The acute phase is generally defined as the first week post-TBI before transitioning into the secondary injury phase that can last months to years [7][10]. Some also advocate for a “subacute” phase as an intermediary that occurs up to 3 months post-TBI [11]. In any case, oxidative stress seems to be a key mediator in the secondary injury phase, as glutamate excitotoxicity, mitochondria dysfunction, and endoplasmic reticulum (ER) stress all contribute to increased reactive oxygen species (ROS) [12][13][14]. Depolarization of glutaminergic neurons after TBI results in increased calcium ion influx through NMDA and AMPA receptors [15]. Excess intracellular calcium increases mitochondrial ROS production through several mechanisms including activation of Ca2+-calmodulin pathways and disruption of the electron transport chain [16]. Endogenous oxidative stress responses are coordinated by the transcription factor Nrf2 [17]. Nrf2 promotes the expression of many cytoprotective proteins including HO-1, NQO-1, and GCLM, among others. These systems can be overwhelmed and become insufficient to prevent ROS-mediated cellular injury [18]. ROS can cause protein damage and misfolding (discussed further below) but may also be especially harmful through lipid peroxidation [19]. Dysregulation of membrane structures such as caveolae in mice is associated with increased markers of neurodegeneration and neuroinflammation [20]. Increased tissue markers of oxidative stress including lipid peroxidation have been observed as far as 12 weeks post-TBI in rats, indicating these pathological mechanisms do not resolve in the acute phase after TBI [21].
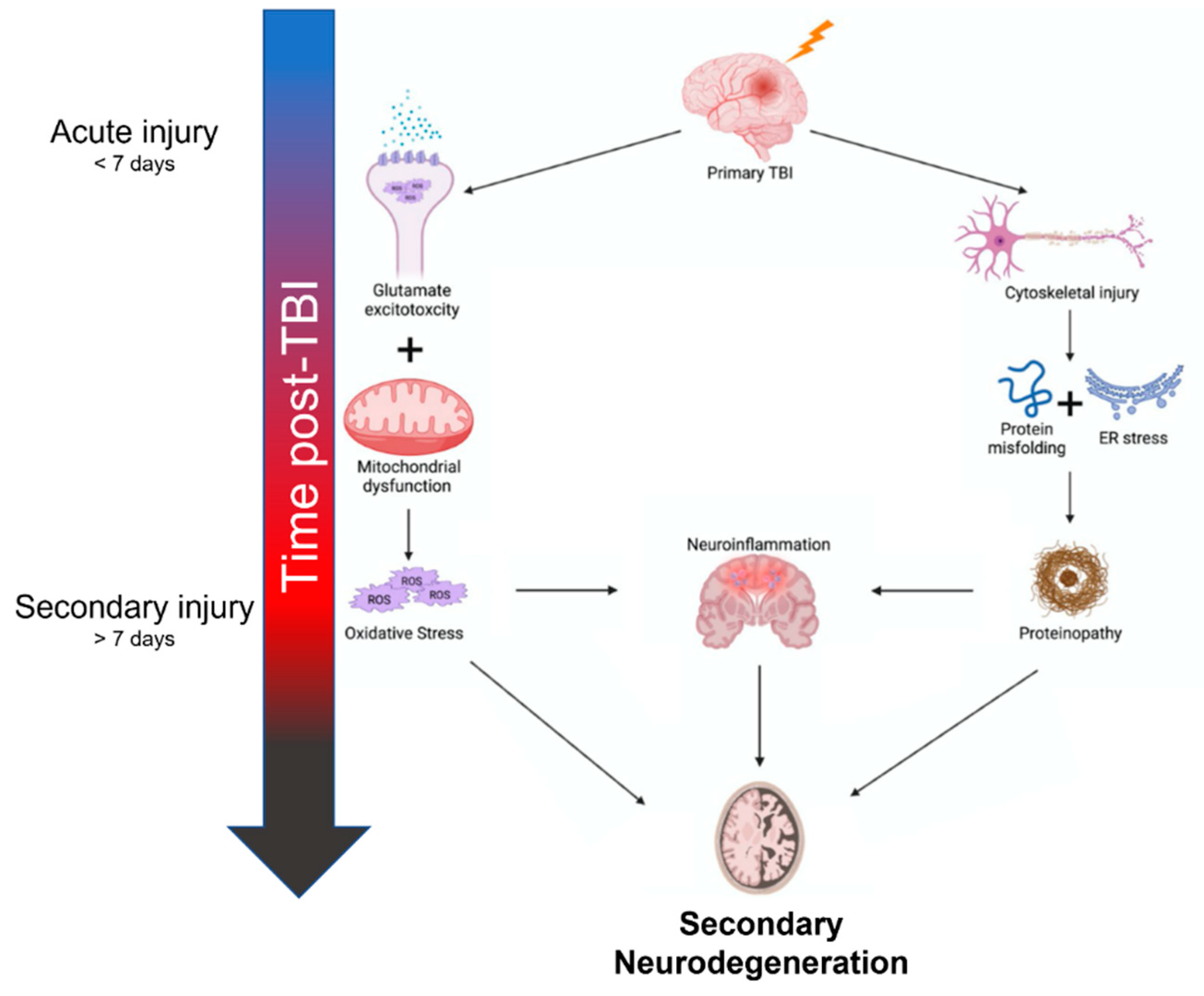
Figure 1. Summary of mechanisms contributing to secondary neurodegeneration following traumatic brain injury.
Another central element of secondary neurodegeneration is pathological changes to neuronal cytoskeletal dynamics. Axons, especially within the white matter, are particularly susceptible to damage from tensile strain during traumatic injuries due to their unique cellular anatomy [22]. The cytoskeleton of these axons can be completely severed during trauma; however, axonal transport may be disrupted even with mild cytoskeletal damage [22]. This type of injury, often termed diffuse axonal injury (DAI) is common after TBI; however, is likely underreported due to the limitations of imaging techniques and the inability to perform brain biopsies in this patient population [23]. Disrupted axonal transport is one of several mechanisms that impede neuronal homeostatic mechanisms and lead to activation of neuroinflammatory pathways (NFκB– and inflammasome-mediated increases in IL-1β, IL-6, TNFα, etc.) and cell death (caspase-3-mediated apoptosis) [24][25][26]. Pro-inflammatory signals begin locally in damaged neurons but quickly promote reactive gliosis and widespread propagation of the neuroinflammatory cascades by microglia and astrocytes [10][24]. Vascular tissues affected by ROS and pro-inflammatory cytokines are at risk of defective autoregulatory function which can decrease cerebral blood flow and compound cerebral injury [10][25]. These pathological changes result in high protein turnover, particularly in neurons, and may alter ER function by stressing proteostatic mechanisms [27]. ER stress, particularly activation of the unfolded protein response (UPR), is a critical mediator of neurodegenerative change [12][28]. Proteinopathies that occur as a result of misfolding including tauopathies, amyloid plaques, Lewy bodies, and TDP-43 have all been observed after TBI [29][30][31]. Severe (i.e., associated with >1 h loss of consciousness) TBI triples the risk of developing of Lewy bodies in the substantia nigra [3]. ROS can be both a product and contributing factor to axonal degeneration and neuroinflammation, highlighting the interconnections between mechanisms of secondary neurodegeneration.
3. Imaging
Neuroimaging can be used to identify chronic pathological changes from TBI in addition to the acute injury. Generally, TBI disrupts white matter connections and results in cerebral atrophy [32]. This finding tends to be worst in frontotemporal and limbic areas [33], possibly due to trends in traumatic injury mechanisms [34]. Serial quantitative T1 magnetic resonance imaging (MRI) can evaluate neurodegeneration after TBI in a sensitive but non-specific way by assessing cerebral atrophy and volume loss. An increasing ventricle-to-brain ratio is associated with chronic cerebral atrophy in those with TBI after resolution of the acute phase (weeks to months) [35][36]. Generally, TBI-induced neurodegeneration leads to cerebral volumes comparable to that of older individuals with other neurodegenerative diseases; both demonstrating a yearly loss of 1.5% of cerebral volume occurring mostly in sulci and white matter tracts [32][37]. The frontotemporal and limbic areas, which are seated on the sharp sphenoid ridge and edge of the tentorium cerebelli, demonstrate the most severe degenerative changes as their location makes them vulnerable to mechanical deformation. Hippocampal atrophy is especially evident within the limbic system considering its location in the medial temporal lobe and high metabolic demand [38][39]. Patients with DAI-type TBI experience white matter degeneration for months to years following the acute injury as evidenced by studies utilizing MRI diffusor tensor imaging (DTI). DTI is a method for detecting structural changes by analyzing the fractional anisotropy (FA), mean diffusivity (MD), and radial diffusivity (RD) of water molecules. TBI with predominant axonal/white matter injury demonstrates reduced FA and increased MD and RD [40][41]. These findings point to demyelination, loss of axonal integrity, and reduced axonal packing and coherence in frontotemporal and limbic structures such as the anterior limb of the internal capsule, corona radiata, optic radiations, and cingulum [42][43]. Furthermore, changes in these DTI indices are associated with poor neuropsychological performance, including executive function, memory, and functional outcomes [44][45][46][47].
Molecular imaging with positron emission tomography is an evolving modality to identify post-traumatic neurodegeneration in vivo in a specific manner. Following TBI, the PET tracer 11C-Pittsburgh compound-B (11C-PiB) binds amyloid-beta (Aβ) in cortical areas, the striatum, and posterior cingulate cortex similar to Alzheimer’s disease (AD). Unlike AD, there are increased Aβ depositions in the cerebellum in TBI [48][49][50]. Additionally, several PET tracers specific to hyperphosphorylated tau (p-tau) previously used in AD are under investigation for use in TBI. FDDNP is the most well-studied biomarker. FDDNP levels are increased in the midbrain, thalamus, pons, and cingulate gyrus and demonstrate lower binding in temporal and parietal regions in military personnel with mild TBI exposure and football players with suspected chronic traumatic encephalopathy compared to patients with AD [51]; however, FDDNP is non-specific as it also binds Aβ. Other PET imaging biomarkers that bind tau include T801, AV1451, and flortaucipir. Studies are generally limited as they have small sample sizes, lack control groups, or are restricted to one subtype of TBI. While cortical tau tracer uptake varies within individuals with CTE-type TBI, studies have demonstrated consistent uptake in the temporal lobe and limbic system [47][52][53][54]. The current use of PET imaging biomarkers remains in the early stages. The Enhancing Neuroimaging Genetics through Meta-Analysis (ENIGMA) consortium is evaluating the efficacy of these imaging modalities alone and in combination with fluid biomarkers, radiogenomics, or with EEG. Furthermore, there is evolving research investigating magnetic resonance spectroscopy (MRS), functional MRI (fMRI), transcranial Doppler (TCD), single photon emission computed tomography (SPECT), and functional near-infrared spectroscopy) [53][55][56]. Early human and rodent studies evaluating the newly discovered glymphatic pathway reveal that mild, repetitive TBI alters glymphatic clearance rates examined with MRI [57][58][59][60].
References
- Fesharaki-Zadeh, A. Chronic Traumatic Encephalopathy: A Brief Overview. Front. Neurol. 2019, 10, 713.
- Raymont, V.; Thayanandan, T. What do we know about the risks of developing dementia after traumatic brain injury? Minerva Med. 2021, 112, 288–297.
- Crane, P.K.; Gibbons, L.E.; Dams-O’Connor, K.; Trittschuh, E.; Leverenz, J.B.; Keene, C.D.; Sonnen, J.; Montine, T.J.; Bennett, D.A.; Leurgans, S.; et al. Association between Traumatic Brain Injury and Late Life Neurodegenerative Conditions and Neuropathological Findings. JAMA Neurol. 2016, 73, 1062–1069.
- Delic, V.; Beck, K.D.; Pang, K.C.H.; Citron, B.A. Biological links between traumatic brain injury and Parkinson’s disease. Acta Neuropathol. Commun. 2020, 8, 45.
- Ramos-Cejudo, J.; Wisniewski, T.; Marmar, C.; Zetterberg, H.; Blennow, K.; de Leon, M.J.; Fossati, S. Traumatic Brain Injury and Alzheimer’s Disease: The Cerebrovascular Link. eBioMedicine 2018, 28, 21–30.
- VanItallie, T.B. Traumatic brain injury (TBI) in collision sports: Possible mechanisms of transformation into chronic traumatic encephalopathy (CTE). Metabolism 2019, 100, 153943.
- Werner, C.; Engelhard, K. Pathophysiology of traumatic brain injury. Br. J. Anaesth. 2007, 99, 4–9.
- Gaetz, M. The neurophysiology of brain injury. Clin. Neurophysiol. 2004, 115, 4–18.
- Johnstone, V.P.; Shultz, S.R.; Yan, E.B.; O’Brien, T.J.; Rajan, R. The acute phase of mild traumatic brain injury is characterized by a distance-dependent neuronal hypoactivity. J. Neurotrauma. 2014, 31, 1881–1895.
- Risbrough, V.B.; Vaughn, M.N.; Friend, S.F. Role of Inflammation in Traumatic Brain Injury–Associated Risk for Neuropsychiatric Disorders: State of the Evidence and Where Do We Go from Here. Biol. Psychiatry 2022, 91, 438–448.
- Mayer, A.R.; Quinn, D.K.; Master, C.L. The spectrum of mild traumatic brain injury: A review. Neurology 2017, 89, 623–632.
- Hetz, C.; Saxena, S. ER stress and the unfolded protein response in neurodegeneration. Nat. Rev. Neurol. 2017, 13, 477–491.
- Freire, M.A.M. Pathophysiology of neurodegeneration following traumatic brain injury. West Indian Med. J. 2012, 61, 751–755.
- Cruz-Haces, M.; Tang, J.; Acosta, G.; Fernandez, J.; Shi, R. Pathological correlations between traumatic brain injury and chronic neurodegenerative diseases. Transl. Neurodegener. 2017, 6, 20.
- Khatri, N.; Thakur, M.; Pareek, V.; Kumar, S.; Sharma, S.; Datusalia, A.K. Oxidative Stress: Major Threat in Traumatic Brain Injury. CNS Neurol. Disord. Drug Targets 2018, 17, 689–695.
- Peng, T.-I.; Jou, M.-J. Oxidative stress caused by mitochondrial calcium overload. Ann. N. Y. Acad. Sci. 2010, 1201, 183–188.
- Ma, Q. Role of Nrf2 in Oxidative Stress and Toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426.
- Zhang, L.; Wang, H. Targeting the NF-E2-Related Factor 2 Pathway: A Novel Strategy for Traumatic Brain Injury. Mol. Neurobiol. 2018, 55, 1773–1785.
- Egawa, J.; Pearn, M.L.; Lemkuil, B.P.; Patel, P.M.; Head, B.P. Membrane lipid rafts and neurobiology: Age-related changes in membrane lipids and loss of neuronal function. J. Physiol. 2016, 594, 4565–4579.
- Head, B.P.; Peart, J.N.; Panneerselvam, M.; Yokoyama, T.; Pearn, M.L.; Niesman, I.R.; Bonds, J.A.; Schilling, J.M.; Miyanohara, A.; Headrick, J.; et al. Loss of Caveolin-1 Accelerates Neurodegeneration and Aging. PLoS ONE 2010, 5, e15697.
- Webster, K.M.; Wright, D.; Sun, M.; Semple, B.D.; Ozturk, E.; Stein, D.G.; O’Brien, T.; Shultz, S.R. Progesterone treatment reduces neuroinflammation, oxidative stress and brain damage and improves long-term outcomes in a rat model of repeated mild traumatic brain injury. J. Neuroinflamm. 2015, 12, 238.
- Johnson, V.E.; Stewart, W.; Smith, D.H. Axonal pathology in traumatic brain injury. Exp. Neurol. 2013, 246, 35–43.
- Jang, S.H. Diagnostic Problems in Diffuse Axonal Injury. Diagnostics 2020, 10, 117.
- Lozano, D.; Schimmel, S.J.; Acosta, S. Neuroinflammation in traumatic brain injury: A chronic response to an acute injury. Brain Circ. 2017, 3, 135–142.
- Pearn, M.L.; Niesman, I.R.; Egawa, J.; Sawada, A.; Almenar-Queralt, A.; Shah, S.B.; Duckworth, J.L.; Head, B.P. Pathophysiology Associated with Traumatic Brain Injury: Current Treatments and Potential Novel Therapeutics. Cell. Mol. Neurobiol. 2017, 37, 571–585.
- O’Brien, W.T.; Pham, L.; Symons, G.F.; Monif, M.; Shultz, S.R.; McDonald, S.J. The NLRP3 inflammasome in traumatic brain injury: Potential as a biomarker and therapeutic target. J. Neuroinflamm. 2020, 17, 104–112.
- Gerakis, Y.; Hetz, C. Emerging roles of ER stress in the etiology and pathogenesis of Alzheimer’s disease. FEBS J. 2018, 285, 995–1011.
- Shah, S.Z.A.; Zhao, D.; Khan, S.H.; Yang, L. Unfolded Protein Response Pathways in Neurodegenerative Diseases. J. Mol. Neurosci. 2015, 57, 529–537.
- Washington, P.M.; Villapol, S.; Burns, M.P. Polypathology and Dementia After Brain Trauma: Does Brain Injury Trigger Distinct Neurodegenerative Diseases, or Should They Be Classified Together as Traumatic Encephalopathy? Exp. Neurol. 2016, 275 Pt 3, 381–388.
- Uryu, K.; Chen, X.-H.; Martinez, D.; Browne, K.D.; Johnson, V.E.; Graham, D.I.; Lee, V.M.-Y.; Trojanowski, J.Q.; Smith, D.H. Multiple proteins implicated in neurodegenerative diseases accumulate in axons after brain trauma in humans. Exp. Neurol. 2007, 208, 185–192.
- Johnson, V.E.; Stewart, W.; Smith, D.H. Widespread τ and amyloid-β pathology many years after a single traumatic brain injury in humans. Brain Pathol. 2012, 22, 142–149.
- Maxwell, W.L.; MacKinnon, M.-A.; Stewart, J.E.; Graham, D.I. Stereology of cerebral cortex after traumatic brain injury matched to the Glasgow Outcome Score. Brain 2010, 133, 139–160.
- Adams, J.H.; Doyle, D.; Ford, I.; Gennarelli, T.A.; Graham, D.I.; McLellan, D.R. Diffuse axonal injury in head injury: Definition, diagnosis and grading. Histopathology 1989, 15, 49–59.
- Hofman, P.A.; Verhey, F.R.; Wilmink, J.T.; Rozendaal, N.; Jolles, J. Brain lesions in patients visiting a memory clinic with postconcussional sequelae after mild to moderate brain injury. J. Neuropsychiatry Clin. Neurosci. 2002, 14, 176–184.
- Bigler, E.D.; Tate, D.F. Brain volume, intracranial volume, and dementia. Investig. Radiol. 2001, 36, 539–546.
- Mamere, A.E.; Saraiva, L.A.L.; Matos, A.L.M.; Carneiro, A.A.O.; Santos, A.C. Evaluation of Delayed Neuronal and Axonal Damage Secondary to Moderate and Severe Traumatic Brain Injury Using Quantitative MR Imaging Techniques. Am. J. Neuroradiol. 2009, 30, 947–952.
- Graham, N.S.; Sharp, D.J. Understanding neurodegeneration after traumatic brain injury: From mechanisms to clinical trials in dementia. J. Neurol. Neurosurg. Psychiatry 2019, 90, 1221–1233.
- Bigler, E.D. Anterior and middle cranial fossa in traumatic brain injury: Relevant neuroanatomy and neuropathology in the study of neuropsychological outcome. Neuropsychology 2007, 21, 515–531.
- De La Plata, C.D.M.; Garces, J.; Kojori, E.S.; Grinnan, J.; Krishnan, K.; Pidikiti, R.; Spence, J.; Devous, M.D.; Moore, C.; McColl, R.; et al. Deficits in Functional Connectivity of Hippocampal and Frontal Lobe Circuits After Traumatic Axonal Injury. Arch. Neurol. 2011, 68, 74–84.
- Veeramuthu, V.; Narayanan, V.; Kuo, T.L.; Delano-Wood, L.; Chinna, K.; Bondi, M.W.; Waran, V.; Ganesan, D.; Ramli, N. Diffusion Tensor Imaging Parameters in Mild Traumatic Brain Injury and Its Correlation with Early Neuropsychological Impairment: A Longitudinal Study. J. Neurotrauma 2015, 32, 1497–1509.
- Farbota, K.D.; Bendlin, B.B.; Alexander, A.L.; Rowley, H.A.; Dempsey, R.J.; Johnson, S.C. Longitudinal diffusion tensor imaging and neuropsychological correlates in traumatic brain injury patients. Front. Hum. Neurosci. 2012, 6, 160.
- Hellyer, P.J.; Leech, R.; Ham, T.E.; Bonnelle, V.; Sharp, D.J. Individual prediction of white matter injury following traumatic brain injury. Ann. Neurol. 2013, 73, 489–499.
- Holleran, L.; Kim, J.H.; Gangolli, M.; Stein, T.; Alvarez, V.; McKee, A.; Brody, D.L. Axonal disruption in white matter underlying cortical sulcus tau pathology in chronic traumatic encephalopathy. Acta Neuropathol. 2017, 133, 367–380.
- Palacios, E.M.; Sala-Llonch, R.; Junque, C.; Roig, T.; Tormos, J.M.; Bargallo, N.; Vendrell, P. White matter integrity related to functional working memory networks in traumatic brain injury. Neurology 2012, 78, 852–860.
- Wang, J.Y.; Bakhadirov, K.; Abdi, H.; Devous, M.D.; De La Plata, C.D.M.; Moore, C.; Madden, C.J.; Diaz-Arrastia, R. Longitudinal changes of structural connectivity in traumatic axonal injury. Neurology 2011, 77, 818–826.
- Sidaros, A.; Engberg, A.W.; Sidaros, K.; Liptrot, M.G.; Herning, M.; Petersen, P.; Paulson, O.B.; Jernigan, T.L.; Rostrup, E. Diffusion tensor imaging during recovery from severe traumatic brain injury and relation to clinical outcome: A longitudinal study. Brain 2008, 131, 559–572.
- Stern, R.A.; Adler, C.H.; Chen, K.; Navitsky, M.; Luo, J.; Dodick, D.W.; Alosco, M.L.; Tripodis, Y.; Goradia, D.D.; Martin, B.; et al. Tau Positron-Emission Tomography in Former National Football League Players. N. Engl. J. Med. 2019, 380, 1716–1725.
- Hong, Y.T.; Veenith, T.; Dewar, D.; Outtrim, J.G.; Mani, V.; Williams, C.; Pimlott, S.; Hutchinson, P.J.; Tavares, A.; Canales, R.; et al. Amyloid imaging with carbon 11-labeled Pittsburgh compound B for traumatic brain injury. JAMA Neurol. 2014, 71, 23–31.
- Mielke, M.M.; Savica, R.; Wiste, H.J.; Weigand, S.D.; Vemuri, P.; Knopman, D.S.; Lowe, V.J.; Roberts, R.O.; Machulda, M.M.; Geda, Y.E.; et al. Head trauma and in vivo measures of amyloid and neurodegeneration in a population-based study. Neurology 2014, 82, 70–76.
- Scott, G.; Ramlackhansingh, A.F.; Edison, P.; Hellyer, P.; Cole, J.; Veronese, M.; Leech, R.; Greenwood, R.J.; Turkheimer, F.E.; Gentleman, S.M.; et al. Amyloid pathology and axonal injury after brain trauma. Neurology 2016, 86, 821–828.
- Chen, S.T.; Siddarth, P.; Merrill, D.A.; Martinez, J.; Emerson, N.D.; Liu, J.; Wong, K.-P.; Satyamurthy, N.; Giza, C.C.; Huang, S.-C.; et al. FDDNP-PET Tau Brain Protein Binding Patterns in Military Personnel with Suspected Chronic Traumatic Encephalopathy1. J. Alzheimer’s Dis. 2018, 65, 79–88.
- Lee, B.; Leavitt, M.J.; Bernick, C.B.; Leger, G.C.; Rabinovici, G.; Banks, S.J. A Systematic Review of Positron Emission Tomography of Tau, Amyloid Beta, and Neuroinflammation in Chronic Traumatic Encephalopathy: The Evidence to Date. J. Neurotrauma 2018, 35, 2015–2024.
- Pierre, K.; Dyson, K.; Dagra, A.; Williams, E.; Porche, K.; Lucke-Wold, B. Chronic Traumatic Encephalopathy: Update on Current Clinical Diagnosis and Management. Biomedicines 2021, 9, 415.
- Barrio, J.R.; Small, G.W.; Wong, K.P.; Huang, S.C.; Liu, J.; Merrill, D.A.; Giza, C.C.; Fitzsimmons, R.P.; Omalu, B.; Bailes, J.; et al. In Vivo characterization of chronic traumatic encephalopathy using FDDNP PET brain imaging. Proc. Natl. Acad. Sci. USA 2015, 112, E2039–E2047.
- Olsen, A.; Babikian, T.; Bigler, E.D.; Caeyenberghs, K.; Conde, V.; Dams-O’Connor, K.; Dobryakova, E.; Genova, H.; Grafman, J.; Håberg, A.K.; et al. Toward a global and reproducible science for brain imaging in neurotrauma: The ENIGMA adult moderate/severe traumatic brain injury working group. Brain Imaging Behav. 2021, 15, 526–554.
- Amyot, F.; Arciniegas, D.B.; Brazaitis, M.P.; Curley, K.C.; Diaz-Arrastia, R.; Gandjbakhche, A.; Herscovitch, P.; Hinds, S.R., 2nd; Manley, G.T.; Pacifico, A.; et al. A Review of the Effectiveness of Neuroimaging Modalities for the Detection of Traumatic Brain Injury. J. Neurotrauma 2015, 32, 1693–1721.
- Christensen, J.; Wright, D.K.; Yamakawa, G.R.; Shultz, S.R.; Mychasiuk, R. Repetitive Mild Traumatic Brain Injury Alters Glymphatic Clearance Rates in Limbic Structures of Adolescent Female Rats. Sci. Rep. 2020, 10, 6254.
- Piantino, J.; Schwartz, D.L.; Luther, M.; Newgard, C.; Silbert, L.; Raskind, M.; Pagulayan, K.; Kleinhans, N.; Iliff, J.; Peskind, E. Link between Mild Traumatic Brain Injury, Poor Sleep, and Magnetic Resonance Imaging: Visible Perivascular Spaces in Veterans. J. Neurotrauma 2021, 38, 2391–2399.
- Kaur, J.; Davoodi-Bojd, E.; Fahmy, L.M.; Zhang, L.; Ding, G.; Hu, J.; Zhang, Z.; Chopp, M.; Jiang, Q. Magnetic Resonance Imaging and Modeling of the Glymphatic System. Diagnostics 2020, 10, 344.
- Taoka, T.; Naganawa, S. Glymphatic imaging using MRI. J. Magn. Reson. Imaging 2020, 51, 11–24.
More
Information
Subjects:
Neurosciences
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.4K
Revisions:
2 times
(View History)
Update Date:
11 Oct 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No