Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Brandon Lucke-Wold and Version 2 by Conner Chen.
Traumatic brain injury (TBI) is a devastating event with severe long-term complications. TBI and its sequelae are one of the leading causes of death and disability in those under 50 years old. It is clear that neurotrauma can incite chronic neurodegenerative processes. Chronic traumatic encephalopathy, Parkinson’s disease, and many other neurodegenerative syndromes have all been associated with a history of traumatic brain injury.
- neurotrauma
- secondary mechanisms
- neurodegeneration
1. Introduction
Neurodegeneration following traumatic brain injury is a complex process that is initiated by several distinct pathways which overwhelm homeostatic stress responses and trigger cellular degeneration and death. Recent studies have demonstrated a progression of neurodegenerative processes months and even years after traumatic brain injury, termed secondary neurodegeneration. Secondary neurodegeneration can manifest in many ways depending on specific etiology and affected neuroanatomy. Chronic traumatic encephalopathy (CTE) is a well-known disease closely associated with repeated traumatic brain injuries (TBI) [1][2][1,2]. Parkinson’s disease (PD), frontotemporal dementia (FTD), and other neurodegenerative diseases are less common but can also be induced as a consequence of TBI [3][4][5][3,4,5]. The precise incidence of CTE is hard to quantify due to diagnostic limitations; however, it has gained notoriety due to the prominence of repeated mild TBI in professional sports [1][6][1,6]. On the other hand, severe TBI with greater than 1 h loss of consciousness triples the risk of eventually developing PD [3]. Investigation into neurodegenerative disease secondary to TBI is rapidly evolving due to its complex pathophysiology and important public health implications.
2. Mechanisms of Neurodegeneration after TBI
The inciting mechanisms of secondary neurodegeneration after TBI are an interdependent set of pathological changes initiated by the primary traumatic injury (Figure 1). The temporal evolution of brain injury after TBI is multidimensional and complex but can be conceptualized as overlapping phases. The “acute injury” phase after TBI is characterized by the predominance of mechanical damage resulting from the initial trauma while the “secondary injury” phase is characterized by the delayed emergence of dysregulated metabolism and inflammation pathways [7][8][9][7,8,9]. The acute phase is generally defined as the first week post-TBI before transitioning into the secondary injury phase that can last months to years [7][10][7,10]. Some also advocate for a “subacute” phase as an intermediary that occurs up to 3 months post-TBI [11]. In any case, oxidative stress seems to be a key mediator in the secondary injury phase, as glutamate excitotoxicity, mitochondria dysfunction, and endoplasmic reticulum (ER) stress all contribute to increased reactive oxygen species (ROS) [12][13][14][12,13,14]. Depolarization of glutaminergic neurons after TBI results in increased calcium ion influx through NMDA and AMPA receptors [15]. Excess intracellular calcium increases mitochondrial ROS production through several mechanisms including activation of Ca2+-calmodulin pathways and disruption of the electron transport chain [16]. Endogenous oxidative stress responses are coordinated by the transcription factor Nrf2 [17]. Nrf2 promotes the expression of many cytoprotective proteins including HO-1, NQO-1, and GCLM, among others. These systems can be overwhelmed and become insufficient to prevent ROS-mediated cellular injury [18]. ROS can cause protein damage and misfolding (discussed further below) but may also be especially harmful through lipid peroxidation [19]. Dysregulation of membrane structures such as caveolae in mice is associated with increased markers of neurodegeneration and neuroinflammation [20]. Increased tissue markers of oxidative stress including lipid peroxidation have been observed as far as 12 weeks post-TBI in rats, indicating these pathological mechanisms do not resolve in the acute phase after TBI [21].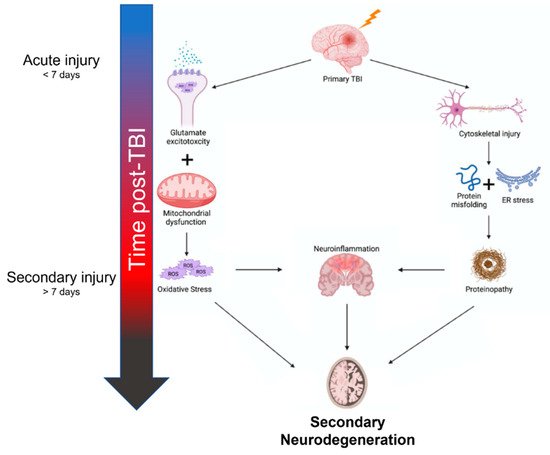
Figure 1.
Summary of mechanisms contributing to secondary neurodegeneration following traumatic brain injury.