 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Chengfeng Yang | + 3563 word(s) | 3563 | 2020-11-02 03:48:01 | | | |
| 2 | Bruce Ren | Meta information modification | 3563 | 2020-11-03 02:23:46 | | | | |
| 3 | Bruce Ren | Meta information modification | 3563 | 2020-11-03 02:28:36 | | | | |
| 4 | Bruce Ren | Meta information modification | 3563 | 2020-11-03 02:43:14 | | |
Video Upload Options
The small Rho GTPases regulate important cellular processes that affect cancer metastasis, such as cell survival and proliferation, actin dynamics, adhesion, migration, invasion and transcriptional activation. The Rho GTPases function as molecular switches cycling between an active GTP-bound and inactive guanosine diphosphate (GDP)-bound conformation. It is known that Rho GTPase activities are mainly regulated by guanine nucleotide exchange factors (RhoGEFs), GTPase-activating proteins (RhoGAPs), GDP dissociation inhibitors (RhoGDIs) and guanine nucleotide exchange modifiers (GEMs). These Rho GTPase regulators are often dysregulated in cancer; however, the underlying mechanisms are not well understood. MicroRNAs (miRNAs), a large family of small non-coding RNAs that negatively regulate protein-coding gene expression, have been shown to play important roles in cancer metastasis. Recent studies showed that miRNAs are capable of directly targeting RhoGAPs, RhoGEFs, and RhoGDIs, and regulate the activities of Rho GTPases. This not only provides new evidence for the critical role of miRNA dysregulation in cancer metastasis, it also reveals novel mechanisms for Rho GTPase regulation.
1. Introduction
Cancer progression is highlighted by changes in cancer cells that promote aggressiveness allowing cells to acquire a greater metastatic potential. Once cancer cells in the primary tumor gain the ability to invade the surrounding tissue, motile cells pass through the basement membrane and the extracellular matrix (ECM) penetrating into the lymphatic or vascular circulation. These motile cells travel through the circulatory system until they arrest at a different locations, extravasate through the vascular basement membrane and the ECM into the new environment where they gain epithelial characteristics and form a secondary or metastatic lesion. Because metastasis is the leading cause of mortality in cancer patients, recent research has focused on identifying and understanding the underlying mechanisms that contribute to metastasis. Numerous studies demonstrated that small Rho GTPases are key regulators of cell adhesion, migration and invasion, thus playing crucial roles in cancer metastasis (for reviews see [1][2][3]). It is well established that the activities of small Rho GTPases are tightly regulated mainly by the following four groups of regulators: guanine nucleotide exchange factors (GEFs), GTPase-activating proteins (GAPs), guanosine diphosphate (GDP) dissociation inhibitors (GDIs) and guanine nucleotide exchange modifiers (GEMs) [4][5][6][7]. However, much less is known about how the activities of small Rho GTPase regulators are regulated.
Although elucidating the underlying mechanisms of cancer metastasis has been the focus for many years, the connection between microRNAs (miRNAs), a family of small non-coding RNAs, and Rho GTPase regulators has only recently become a focused topic in cancer metastasis studies. There is a growing body of evidence revealing the critical involvement of miRNAs in the tight spatiotemporal regulation of actin-based physiology. Moreover, depending on the specific context, miRNAs can have a tumor suppressive or oncogenic role in cancer. We understand that miRNAs can directly regulate the expression of Rho GTPases and this was reviewed elsewhere [8]. In this review, we focused on recent exciting findings showing that miRNAs play important roles in regulating Rho GTPase regulators (RhoGEFs, RhoGAPs, RhoGDIs), eventually affecting small Rho GTPase activities and cancer metastasis. A comprehensive list of the currently validated miRNA-targeting of small Rho GTPase regulators is presented.
2. MicroRNA Biogenesis and Function
Although the basic features of microRNA biogenesis and its mechanism of action were established over a decade ago [9][10][11], subsequent years have shown a vast accumulation of new information that has not only deciphered the mechanistic details, but has also demonstrated that miRNAs are key regulatory hubs for cancer. Here, we provide only a brief introduction to miRNA biogenesis and function for context as we discuss their direct role in modulating mechanisms that contribute to cancer progression (we have previously reviewed miRNA biogenesis in more detail [12][13][14]).
MicroRNAs (miRNAs or miRs), are a subclass of small (~21–23 nucleotides) non-coding RNA molecules that negatively regulate protein-coding gene expression. In terms of biogenesis (Figure 1), a functionally mature miRNA is derived from the cleavage of a double-stranded ~70 nt RNA hairpin precursor in the cytosol. These miRNA precursors are typically located either within the introns of a host protein-coding gene or in intergenic regions, and are transcribed in the nucleus by either RNA polymerase II or III. However, the cases in which miRNA precursors were found within the exons of transcripts and in antisense transcripts have been reported [15][16]. Once excised from the precursor RNA hairpin, a mature miRNA is then loaded into the RNA-induced silencing complex (RISC), where miRNAs are then able to negatively regulate the expression of target genes. Functionally, miRNAs elicit this negative regulation typically by imperfect base pairing with the 3′ untranslated region (3′UTR) of the target messenger RNA (mRNA) through the miRNA seed sequence. The seed sequence is the second to eighth nucleotide at the 5′ end of a mature miRNA. The binding of a miRNA to its target mRNAs can induce mRNA degradation, translational inhibition or direct cleavage, depending on the sequence complementarity [17][18][19]. The expression of miRNAs can be regulated through interactions with and the modifications of their promoters. In addition to the promoter-mediated control of expression, miRNA function can be controlled through the binding and sequestration of the mature miRNA in the cytosol by long non-coding RNAs (lncRNAs) as well as circular ncRNAs (circRNAs), termed competing endogenous RNAs (ceRNAs) [20]. Due to their direct role in regulating the gene expression of most of the human genome[21][22], miRNAs are directly involved in almost all aspects of cellular functions. Specifically, the cellular pathways that underlie cancer progression are regulated by either oncogenic or tumor suppressive miRNAs [23]. This suggests that miRNAs are central regulatory elements and represent a promising avenue for therapeutic intervention.
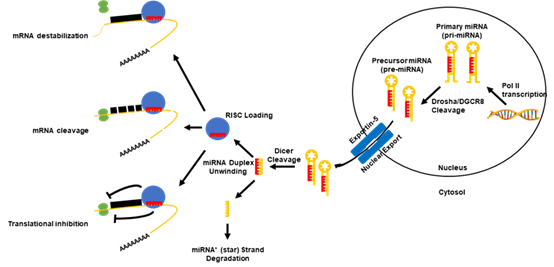
Figure 1. Canonical microRNA (miRNA) biogenesis pathway. Canonically, the miRNAs are transcribed in the nucleus via their own promoters or their host gene promoters by RNA polymerase II or III. This results in the formation of a primary miRNA (pri-miRNA) transcript, which can range from hundreds to thousands of nucleotides long. After a polyadenylation and capping event, pri-miRNAs undergo a microprocessing cleavage event by a ribonuclease (RNase) III type enzyme, Drosha, and its binding partner DiGeorge syndrome critical region gene 8 (DGCR8) resulting in a ~60–120 nucleotide-long precursor miRNA transcript (pre-miRNA). Pre-miRNAs are then exported out of the nucleus by the karyopherin exportin-5 to the cytoplasm, where the RNase II enzyme, Dicer, processes the transcripts to form a miRNA duplex. The unwinding of the miRNA duplex occurs and one strand is usually degraded (miRNA* (star) strand), while the other (mature miRNA) is loaded into the RNA-induced silencing complex (RISC). The RISC probes for targets of the miRNA in the genome. Once bound to a target mRNA, the RISC may induce a negative expression of the mRNA in three ways: 1) mRNA destabilization and degradation, 2) mRNA translational inhibition, or 3) mRNA cleavage. The path at which the mRNA is regulated depends upon multiple factors of the mature miRNA.
3. The Small Rho GTPases
Ras small GTPases are a superfamily of monomeric hydrolases that are found in all eukaryotic cells and function similarly to the α-subunits of heterotrimeric G proteins. Small GTPases act as molecular switches to facilitate cell activities including proliferation, morphology change, adhesion, migration, invasion and nuclear or vesicular transport, among others. This molecular switching is driven by binding and hydrolyzing GTP, leading to the transition of small GTPases between three conformational states; 1) the GDP-bound, 2) the GTP-bound and 3) the empty state that transiently exists between the replacement of GDP with GTP in the guanine nucleotide binding site [24]. The three main areas of the GTPase that change between GTP- or GDP-bound states are referred to as the phosphate binding loop (P-loop), switch 1 (residues 30–40, also known as the effector loop) and switch 2 (residues 60–76) [25][26], all of which reside within the GTP-binding site of the GTPase. The GDP-bound state is generally considered inactive (“off”), while the GTP-bound form is the active (“on”) form which allows GTPases to move to the cell membrane region and interact with downstream effectors.
The small Rho GTPase family is one of the five originally classified major subfamilies of the Ras small GTPase superfamily [27]. It consists of 20 small (190–250 residues) molecules which control the cytoskeleton and cell morphology specifically by regulating actin dynamics (Table 1). They share ~30% sequence identity with the other Ras superfamily proteins and between 40–95% sequence identity within the subfamily. In addition to containing sequence motifs common to all Ras small GTPases, what structurally separates Rho small GTPases from other proteins in the Ras superfamily is the insertion of 9–12 residues, after residue 122 located between the fifth β-strand and fourth α-helix within the GTPase domain [28][29]. The majority of Rho GTPases undergo C-terminal post-translational modifications by isoprenoid lipids or palmitate fatty acids [30][31], which help localize their subcellular localization and association with membranes or organelles. In addition to modifications to their C-terminal, Rho GTPases are also directly regulated by GTPase-activating proteins (GAPs), guanine nucleotide exchange factors (GEFs), GDP dissociation inhibitors (GDIs) and guanine nucleotide exchange modifiers (GEMs) discussed later in this review. Of the 20 members of the Rho GTPase subfamily (Table 1), the best characterized Rho molecules are RhoA, Rac1, and Cdc42. RhoA promotes actin–myosin contractility and thus controls stress fiber and focal adhesion formation and turnover. Rac1 drives actin polymerization for the formation of membrane ruffling and lamellipodia, or the large projection at the leading edge of the migrating cell. Cdc42 regulates the formation of filopodia, which are actin-rich, finger-like projections that exude from the lamellipodia at the leading edge of the migrating cell.
Table 1. Mammalian Rho GTPases.
|
GTPase (Alias) |
Function |
Citation |
|
CDC42 (CDC42Hs) |
Transduced signals to the actin cytoskeleton to initiate and maintain polarized growth and mitogen-activate protein morphogenesis; Responsible for the formation of filopodia in actin-based cell migration. |
|
|
RAC1 |
Transduced signals to the actin cytoskeleton to regulate the multiple signaling pathways that control actin cytoskeleton organization, transcription and proliferation; Responsible for the formation of lamellipodia in actin-based cell migration. |
[34] |
|
RAC2 |
Transducer that localized to the plasma membrane, where it worked with Rac1 to regulate diverse processes, such as secretion, phagocytosis and cell polarization; Expressed primarily in hematopoietic cell lineages. |
|
|
RAC3 |
Involved in synaptic potentiation through regulating the actin cytoskeletal dynamics; Primarily expressed in the neurons of ganglia and the central nervous system. |
|
|
RHOA |
Involved in the regulation of cell adhesion and migration; Responsible for providing contractile force in cell migration through the formation of stress fibers and focal adhesions; Localized to the cytoplasm and to a certain degree the plasma membrane. |
|
|
RHOB |
Transducer involved in actin organization, cell migration, membrane and endosome trafficking, proliferation, DNA repair, and apoptosis; Thought to be an inhibitor of cancer progression; Localized to the endosomal membrane. |
[41] |
|
RHOBTB1 |
Function not well known, did not play a major role in the organization of actin cytoskeleton dynamics; Not targetable by RhoGAPs, RhoGEFs, or RhoGDIs; Ubiquitously expressed, although high levels are found in skeletal muscle, placenta, stomach, kidney, testis, adrenal gland and uterus. |
|
|
RHOBTB2 |
Function not well known, did not play a major role in the organization of actin cytoskeleton dynamics; Not targetable by RhoGAPs, RhoGEFs, or RhoGDIs; Weakly expressed, although high levels were found in neural and cardiac tissues. |
|
|
RHOBTB3 |
Function not well known, did not play a major role in the organization of actin cytoskeleton dynamics; Not targetable by RhoGAPs, RhoGEFs, or RhoGDIs; Ubiquitously expressed, although high levels were found in placenta, testis, pancreas, adrenal and salivary gland, and neural and cardiac tissues. |
|
|
RHOC |
Responsible for actin cytoskeletal reorganization involved in promoting cell migration, proliferation, EMT, invasion, angiogenesis and metastasis; Localized to the cytoplasm and with undefined perinuclear structures. |
[44] |
|
RHOD |
Controlled endocytic vesicle movement, Golgi homeostasis, and promoted actin stress fiber dissociation; Localized to the endosomal membrane. |
[45] |
|
RHOE (RND3) |
Expressed ubiquitously; Inhibited contractility and the subsequent formation of actin stress fibers and focal adhesions; Drove cell rounding. GTPase-deficient, but constitutively bound to GTP in vivo. |
[28] |
|
RHOF (RIF) |
Expressed ubiquitously; Promoted the formation of filopodia. |
[28] |
|
RHOG |
Localized to caveolar vesicles; May have played a role in the inflammatory response; Involved in lamellipodia and filopodia formation, and membrane ruffling. |
[28] |
|
RHOH (TTF) |
Expressed primarily in hematopoietic cell lineages; GTPase-deficient; Overexpression inhibited RAC1, RHOA, and CDC42 signaling; Not targetable by RhoGAPs, RhoGEFs, or RhoGDIs. |
[28] |
|
RHOJ (TCL) |
Localized to the endosomal membrane; Promoted the formation of filopodia; Contributed to the focal adhesion turnover. |
[28] |
|
RHON (RND2) |
Expressed primarily in testis, brain, and liver; GTPase-deficient; Involved in neurite outgrowth and cytokinesis. |
[46] |
|
RHOQ (TC10) |
Localized to the endosomal membrane; Promoted the formation of filopodia; Implicated in receptor trafficking. |
[46] |
|
RHOS (RND1) |
Expressed primarily in adult brain and liver; Inhibited contractility and the subsequent formation of actin stress fibers and focal adhesions; Drove cell rounding; GTPase-deficient. |
[46] |
|
RHOU (WRCH1) |
Critical for Wnt signaling; Worked together with RAC1; Stimulated cell cycle progression; Promoted dissociation of stress fibers. |
[28] |
|
RHOV (WRCH2) |
Promoted the formation of filopodia; Promoted the dissociation of stress fibers. |
RhoGAPs: Rho GTPase-activating proteins, RhoGEFs: Rho guanine nucleotide exchange factors, RhoGDIs: Rho guanosine diphosphate (GDP) dissociation inhibitors (GDIs).
4. Regulators of the Small Rho GTPases
The “on” and “off” states of the Rho GTPases can be accelerated by the interaction with certain regulators of G-protein signaling (Figure 2) [4][5][6]. GTPase-activating proteins (GAPs) accelerate the Rho GTPases intrinsic phosphatase capability, putting the GTPase into the “off” state. Conversely, guanine nucleotide exchange factors (GEFs) activate Rho GTPases by rapidly exchanging GDP with the GTP. GDP dissociation factors (GDIs) also act to put the Rho GTPases into the “off” state by binding and sequestering Rho GTPases. Since the number of GAPs and GEFs outnumbers the number of Rho GTPases by over 3 to 1, many of these GAPs, GEFs and GDIs target the same Rho GTPase. However, some of these GAPs, GEFs and GDIs have been shown to be specific for a single Rho GTPase over the others. Moreover, these regulators of small G-protein signaling have been shown to be regulated by the small Rho GTPases themselves.
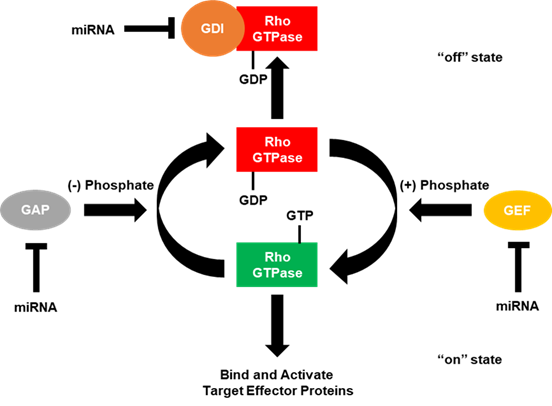
Figure 2. GTPase-activating proteins (GAPs), guanine nucleotide exchange factors (GEFs) and guanosine diphosphate (GDP) dissociation factors (GDIs) regulate small Rho GTPases. Small Rho GTPases are known as molecular switches due to the fact they cycle between “on” (GTP-bound) and “off” (GDP-bound) states. In the GTP-bound state, small Rho GTPases are able to regulate intracellular signaling cascades by binding and activating effector molecules. This signaling can be terminated by the intrinsic GTPase capability (GTP to GDP) of small Rho GTPases, which is enhanced by the interaction with GTPase-activating proteins (GAPs). While in the GDP-bound state, Rho GTPases can also interact with guanosine nucleotide dissociation inhibitors (GDIs), which sequester the small Rho GTPase and do not allow for the GDP to be exchanged for GTP. In order for GDIs to release the small Rho GTPase, a release factor must be present. Conversely, in the GDP-bound state, small Rho GTPases are unable to regulate downstream signaling, but can be reactivated by exchanging GDP for GTP, facilitated by the interaction with a guanine nucleotide exchange factor (GEF). Current literature has demonstrated that miRNAs can directly bind and downregulate the expression of RhoGAPs, GEFs, and GDIs to regulate cancer progression.
4.1. GTPase-Activating Proteins (GAPs)
The Rho GTPase-activating proteins (RhoGAPs) are one of the major regulators of Rho GTPases found in all eukaryotes. They are defined by the presence of a conserved 150 residue RhoGAP domain, which is distinct from GAPs for other classes of GTPases. This domain consists of nine α helices and a conserved arginine residue in a loop structure [47]. The RhoGAP domain gives RhoGAPs their function because it is sufficient for the binding to GTP-bound Rho proteins as well as accelerating their GTPase activity. Traditionally, GAPs were thought of as tumor suppressors. However, as some recent work has demonstrated, the overexpression of RhoGAPs in some cancers [48][49][50] and the interaction between the Rho GTPases and RhoGAPs may be more complex and context-dependent than originally thought. Expanding this to miRNAs, this would also suggest that miRNAs that directly target RhoGAPs also play a context-dependent role in cancer progression.
4.2. Guanine Nucleotide Exchange Factors (GEFs)
Rho guanine nucleotide exchange factors (RhoGEFs) are directly responsible for the activation of Rho GTPases by catalyzing the exchange of GDP for GTP. Some RhoGEFs display specificity toward a single Rho GTPase, while others exhibit more promiscuity. As in the case of RhoGAPs, the highly controlled regulation of RhoGEF activity is paramount to ensure that Rho GTPases are not always in the “on” state. This regulation and specificity of RhoGEFs is determined by regulatory mechanisms such as posttranslational modifications, having differing sensitivities to lipids, direct binding to surface receptors, and an association with specialized complexes [51][52][53]. Based upon their functions, this would suggest that aberrant RhoGEF regulation, such as increased gene expression or mutations causing constitutive activation, is a main driver behind cancer progression. Indeed, RhoGEFs are positive regulators of cancer progression, where their increased expression drives cancer cell migration, invasion, adhesion and metastasis. Therefore, this suggests that miRNAs that directly target RhoGEFs function as tumor suppressors. However, this idea has been challenged recently where a RhoGEF (ARHGEF10) was shown to function as a tumor suppressor in pancreatic ductal adenocarcinoma [54].
There are two subfamilies of RhoGEFs: the diffuse B-cell lymphoma (Dbl) and dedicator of cytokinesis (DOCK) families. The 73 members of the Dbl family (Table 2) share a ~200 residue catalytic Dbl homology (DH) domain immediately preceding an adjacent regulatory ~100 residue pleckstrin homology (PH) domain [55]. However, some family members possess tandem DH and PH domains or completely lack a PH domain [56]. They also differ significantly in the N- and C-terminal sequences, which is used to regulate the intrinsic RhoGEF catalytic activity, localization, or complex association as described above. Additionally, it should be noted that unlike humans, plants do not possess Dbl RhoGEFs [57]. Functionally, the DH domain of Dbl RhoGEFs is responsible for facilitating the exchange of GDP for GTP by stabilizing switch 1, the remodeling of the alanine near the Mg2+ binding site in switch 2, and stabilizing the P-loop of the Rho GTPase. Dbl RhoGEFs have been shown to act on RhoA, Rac1, and Cdc42. Conversely, the DOCK RhoGEFS (Table 2) are characterized by a conserved Dock-homology region-2 (DHR2) domain that serves as the catalytic domain and the DHR1 domain that locates them to specific membranes. What makes these RhoGEFs unique is that the DHR2 domain stabilizes the switch 1 of the Rho GTPase using interactions not seen in other typical RhoGEFs. DOCK proteins also utilize a valine residue to help dissociate the bound GDP, but does not distort switch 2 of the GTPase like Dbl family members . DOCK proteins are shown to act primarily as RhoGEFs for Rac1 and Cdc42, but not RhoA [58][59].
4.3. GDP Dissociation Inhibitors (GDIs)
In addition to RhoGAPs and RhoGEFs, Rho GTPase GDP dissociation inhibitors (RhoGDIs) perform a unique function in the regulation of Rho GTPases. Characterized by a conserved ~60 residue N-terminal domain, RhoGDIs prevent Rho GTPases from activation and the subsequent interactions with downstream effectors through three different mechanisms: 1) binding and sequestering Rho GTPases in the inactive, GDP-bound form preventing activation by RhoGEFs. 2) binding and sequestering Rho GTPases in the active, GTP-bound form preventing the hydrolysis of GTP by either the intrinsic or the RhoGAP-stimulated GTPase activity, and 3) modulating the cycling of Rho GTPases between the cytosolic and membrane localization [60][61]. These biochemical functions mean that RhoGDIs have dual roles in the cell; they form soluble complexes with GDP-bound Rho GTPases in the cytosol, but also monitor Rho GTPases at the site of action on membranes.
To elicit their effects, RhoGDIs recognize the isoprenoid geranylgeranyl lipids at the C-terminus of the Rho GTPase [60][61]. Once bound to the Rho GTPase, the N-terminal domain of the RhoGDI interacts with the switch 1 and switch 2 of the Rho GTPase, restricting the spatial flexibility needed to exchange GDP or hydrolyze GTP. In contrast to the large number of RhoGAPs and RhoGEFs, only three RhoGDIs have been identified in mammals (Table 2). RhoGDIα (also known as RhoGDI1) is the most commonly found and ubiquitously expressed RhoGDI and it is able to form complexes with most members of the Rho family [62]. RhoGDIβ (also known as RhoGDI2) is predominantly expressed in hematopoietic cells [63][64], but its dysregulation is also found in certain cancer types [66][67][68]. It can interact with several Rho GTPases, but the affinity for complexing is 10–20 fold lower than that of RhoGDIα [69]. Lastly, RhoGDIɣ (also known as RhoGDI3) is preferentially expressed in the brain, lung, kidney, testis and pancreas , and is targeted to the Golgi complex through its N-terminal domain where it predominantly interacts with RhoB and RhoG . The dysregulation of the RhoGDIs is linked to cancer cell migration, invasion, and metastasis [70][71], but the downstream effects of the altered RhoGDI expression seems to be context and cancer-type dependent. Additionally, it has been shown that RhoGDI mRNA can interact with regulators critical to the miRNA biogenesis, stability and activity [72], suggesting a more diverse role for RhoGDIs in cancer cells.
4.4. Guanine Nucleotide Exchange Modulators (GEMs)
In addition to GAPs, GEFs, and GDIs, another family of GTPase regulators has recently been identified. The guanine nucleotide exchange modulators (GEMs) are unique because they function as both a GEF and a GDI depending on context [73]. The members of this family are characterized by a ~30 residue domain that directly binds to G proteins. GEMs share little sequence homology between family members and act as central regulators to diverse G protein signaling cascades. The prototypical GEM family member is Girdin (for Girder of actin [74], also known as GIV [75], HkRP1 [76], or APE [77]), which is a multi-domain cytosolic protein that was identified to regulate the actin cytoskeleton during cell migration. Although it has been shown to drive actin cytoskeletal remodeling and bind to α-subunits of heterotrimeric G proteins like other GTPase regulators, these studies were not performed using human Rho GTPases. Therefore, it is not yet known if GEMs can directly modulate Rho GTPases activity.
References
- Ridley, A.J. Rho GTPases and cell migration. J. Cell Sci. 2001, 114, 2713–2722.
- Lawson, C.D.; Ridley, A.J. Rho GTPase signaling complexes in cell migration and invasion. J. Cell Biol. 2018, 217, 447–457, doi:10.1083/jcb.201612069.
- Schmitz, A.A.; Govek, E.E.; Böttner, B.; Van Aelst, L. Rho GTPases: Signaling, migration, and invasion. Exp. Cell Res. 2000, 261, 1–12.
- Kaibuchi, K.; Kuroda, S.; Amano, M. Regulation of the Cytoskeleton and Cell Adhesion by the Rho Family GTPases in Mammalian Cells. Annu. Rev. Biochem. 1999, 68, 459–486.
- Takai, Y.; Sasaki, T.; Matozaki, T. Small GTP-Binding Proteins. Physiol. Rev. 2001, 81, 153–208.
- Cherfils, J.; Zeghouf, M. Regulation of small GTPases by GEFs, GAPs, and GDIs. Physiol. Rev. 2013, 93, 269–309, doi:10.1152/physrev.00003.2012.
- Ghosh, P.; Rangamani, P.; Kufareva, I. The GAPs, GEFs, GDIs and…now, GEMs: New kids on the heterotrimeric G protein signaling block. Cell Cycle 2017, 16, 607–612, doi:10.1080/15384101.2017.1282584.
- Liu, M.; Bi, F.; Zhou, X.; Zheng, Y. Rho GTPase regulation by miRNAs and covalent modifications. Trends Cell Biol. 2012, 22, 365–373, doi:10.1016/j.tcb.2012.04.004.
- Bartel, D.P. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell 2004, 116, 281–297.
- Brennecke, J.; Stark, A.; Russell, R.B.; Cohen, S.M. Principles of microRNA-target recognition. PLoS Biol. 2005, 3, e85, doi:10.1371/journal.pbio.0030085.
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233, doi:10.1016/j.cell.2009.01.002.
- Humphries, B.; Yang, C. The microRNA-200 family: Small molecules with novel roles in cancer development, progression, and therapy. Oncotarget 2015, 6, 6472–6498.
- Humphries, B.; Wang, Z.; Yang, C. MicroRNA Regulation of Epigenetic Modifiers in Breast Cancer. Cancers 2019, 11, doi:10.3390/cancers11070897.
- Xiao, Y.; Humphries, B.; Yang, C.; Wang, Z. MiR-205 Dysregulations in Breast Cancer: The Complexity and Opportunities. Noncoding RNA 2019, 5, doi:10.3390/ncrna5040053.
- Lagos-Quintana, M.; Rauhut, R.; Meyer, J.; Borkhardt, A.; Tuschl, T. New microRNAs from mouse and human. RNA 2003, 9, 175–179, doi:10.1261/rna.2146903.
- Rodriguez, A.; Griffiths-Jones, S.; Ashurst, J.L.; Bradley, A. Identification of mammalian microRNA host genes and transcription units. Genome Res. 2004, 14, 1902–1910, doi:10.1101/gr.2722704.
- Pillai, R.S.; Bhattacharyya, S.N.; Filipowicz, W. Repression of protein synthesis by miRNAs: How many mechanisms? Trends Cell Biol. 2007, 17, 118–126, doi:10.1016/j.tcb.2006.12.007.
- Karginov, F.V.; Cheloufi, S.; Chong, M.M.; Stark, A.; Smith, A.D.; Hannon, G.J. Diverse endonucleolytic cleavage sites in the mammalian transcriptome depend upon microRNAs, Drosha, and additional nucleases. Mol. Cell 2010, 38, 781–788, doi:10.1016/j.molcel.2010.06.001.
- Bracken, C.P.; Szubert, J.M.; Mercer, T.R.; Dinger, M.E.; Thomson, D.W.; Mattick, J.S.; Michael, M.Z.; Goodall, G.J. Global analysis of the mammalian RNA degradome reveals widespread miRNA-dependent and miRNA-independent endonucleolytic cleavage. Nucleic Acids Res. 2011, 39, 5658–5668, doi:10.1093/nar/gkr110.
- Kartha, R.V.; Subramanian, S. Competing endogenous RNAs (ceRNAs): New entrants to the intricacies of gene regulation. Front. Genet. 2014, 5, 8, doi:10.3389/fgene.2014.00008.
- Friedman, R.C.; Farh, K.K.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105, doi:10.1101/gr.082701.108.
- Griffiths-Jones, S.; Saini, H.K.; van Dongen, S.; Enright, A.J. miRBase: Tools for microRNA genomics. Nucleic Acids Res. 2008, 36, D154–D158, doi:10.1093/nar/gkm952.
- Peng, Y.; Croce, C.M. The role of MicroRNAs in human cancer. Signal Transduct. Target. Ther. 2016, 1, 15004, doi:10.1038/sigtrans.2015.4.
- Bourne, H.R.; Sanders, D.A.; McCormick, F. The GTPase superfamily: A conserved switch for diverse cell functions. Nature 1990, 348, 125–132.
- Pai, E.F.; Kabsch, W.; Krengel, U.; Holmes, K.C.; John, J.; Wittinghofer, A. Structure of the guanine-nucleotide-binding domain of the Ha-ras oncogene product p21 in the triphosphate conformation. Nature 1989, 341, 209–214.
- Milburn, M.V.; Tong, L.; DeVos, A.M.; Brünger, A.; Yamaizumi, Z.; Nishimura, S.; Kim, S.H. Molecular Switch for Signal Transduction: Structural Differences Between Active and Inactive Forms of Protooncogenic ras Proteins. Science 1990, 247, 939–945.
- Kahn, R.A.; Der, C.J.; Bokoch, G.M. The ras superfamily of GTP-binding proteins: Guidelines on nomenclature. FASEB J. 1992, 6, 2512–2513.
- Wennerberg, K.; Der, C.J. Rho-family GTPases: it’s not only Rac and Rho (and I like it). J. Cell Sci. 2004, 117, 1301–1312, doi:10.1242/jcs.01118.
- Valencia, A.; Chardin, P.; Wittinghofer, A.; Sander, C. The ras Protein Family: Evolutionary Tree and Role of Conserved Amino Acids. Biochemistry 1991, 30, 4637–4648, doi:10.1021/bi00233a001‚.
- Roberts, P.J.; Mitin, N.; Keller, P.J.; Chenette, E.J.; Madigan, J.P.; Currin, R.O.; Cox, A.D.; Wilson, O.; Kirschmeier, P.; Der, C.J. Rho Family GTPase modification and dependence on CAAX motif-signaled posttranslational modification. J. Biol. Chem. 2008, 283, 25150–25163, doi:10.1074/jbc.M800882200.
- Adamson, P.; Marshall, C.J.; Hall, A.; Tilbrook, P.A. Post-translational Modifications of p21rho Proteins. J. Biol. Chem. 1992, 267, 20033–20038.
- Johnson, D.I. Cdc42: An Essential Rho-Type GTPase Controlling Eukaryotic Cell Polarity. Microbiol. Mol. Biol. Rev. 1999, 63, 54–105.
- Etienne-Manneville, S. Cdc42-the centre of polarity. J. Cell Sci. 2004, 117, 1291–1300, doi:10.1242/jcs.01115.
- Bosco, E.E.; Mulloy, J.C.; Zheng, Y. Rac1 GTPase: A “Rac” of all trades. Cell. Mol. Life Sci. 2009, 66, 370–374, doi:10.1007/s00018-008-8552-x.
- Gu, Y.; Jia, B.; Yang, F.C.; D’Souza, M.; Harris, C.E.; Derrow, C.W.; Zheng, Y.; Williams, D.A. Biochemical and biological characterization of a human Rac2 GTPase mutant associated with phagocytic immunodeficiency. J. Biol. Chem. 2001, 276, 15929–15938, doi:10.1074/jbc.M010445200.
- Troeger, A.; Williams, D.A. Hematopoietic-specific Rho GTPases Rac2 and RhoH and human blood disorders. Exp. Cell Res. 2013, 319, 2375–2383, doi:10.1016/j.yexcr.2013.07.002.
- De Curtis, I. The Rac3 GTPase in Neuronal Development, Neurodevelopmental Disorders, and Cancer. Cells 2019, 8, doi:10.3390/cells8091063.
- Bustelo, X.R.; Sauzeau, V.; Berenjeno, I.M. GTP-binding proteins of the Rho/Rac family: Regulation, effectors and functions in vivo. Bioessays 2007, 29, 356–370.
- Hanna, S.; El-Sibai, M. Signaling networks of Rho GTPases in cell motility. Cell. Signal. 2013, 25, 1955–1961, doi:10.1016/j.cellsig.2013.04.009.
- Sit, S.T.; Manser, E. Rho GTPases and their role in organizing the actin cytoskeleton. J. Cell Sci. 2011, 124, 679–683, doi:10.1242/jcs.064964.
- Vega, F.M.; Ridley, A.J. The RhoB small GTPase in physiology and disease. Small GTPases 2018, 9, 384–393, doi:10.1080/21541248.2016.1253528.
- Ji, W.; Rivero, F. Atypical Rho GTPases of the RhoBTB Subfamily: Roles in Vesicle Trafficking and Tumorigenesis. Cells 2016, 5, doi:10.3390/cells5020028.
- Berthold, J.; Schenkova, K.; Rivero, F. Rho GTPases of the RhoBTB subfamily and tumorigenesis. Acta Pharmacol. Sin. 2008, 29, 285–295, doi:10.1111/j.1745-7254.2008.00773.x.
- Thomas, P.; Pranatharthi, A.; Ross, C.; Srivastava, S. RhoC: A fascinating journey from a cytoskeletal organizer to a Cancer stem cell therapeutic target. J. Exp. Clin. Cancer Res. 2019, 38, 328, doi:10.1186/s13046-019-1327-4.
- Phuyal, S.; Farhan, H. Multifaceted Rho GTPase Signaling at the Endomembranes. Front. Cell Dev. Biol. 2019, 7, 127, doi:10.3389/fcell.2019.00127.
- Vega, F.M.; Ridley, A.J. Rho GTPases in cancer cell biology. FEBS Lett. 2008, 582, 2093–2101, doi:10.1016/j.febslet.2008.04.039.
- Gamblin, S.J.; Smerdon, S.J. GTPase-activating proteins and their complexes. Curr. Opin. Struct. Biol. 1998, 8, 195–201.
- Lazarini, M.; Traina, F.; Machado-Neto, J.A.; Barcellos, K.S.; Moreira, Y.B.; Brandao, M.M.; Verjovski-Almeida, S.; Ridley, A.J.; Saad, S.T. ARHGAP21 is a RhoGAP for RhoA and RhoC with a role in proliferation and migration of prostate adenocarcinoma cells. Biochim. Biophys. Acta 2013, 1832, 365–374, doi:10.1016/j.bbadis.2012.11.010.
- Johnstone, C.N.; Castellvi-Bel, S.; Chang, L.M.; Bessa, X.; Nakagawa, H.; Harada, H.; Sung, R.K.; Pique, J.M.; Castells, A.; Rustgi, A.K. ARHGAP8 is a novel member of the RHOGAP family related to ARHGAP1/CDC42GAP/p50RHOGAP: Mutation and expression analyses in colorectal and breast cancers. Gene 2004, 336, 59–71, doi:10.1016/j.gene.2004.01.025.
- Humphries, B.; Wang, Z.; Li, Y.; Jhan, J.R.; Jiang, Y.; Yang, C. ARHGAP18 Downregulation by miR-200b Suppresses Metastasis of Triple-Negative Breast Cancer by Enhancing Activation of RhoA. Cancer Res. 2017, 77, 4051–4064, doi:10.1158/0008-5472.CAN-16-3141.
- Barrio-Real, L.; Kazanietz, M.G. Rho GEFs and Cancer: Linking Gene Expression and Metastatic Dissemination. Sci. Signal. 2012, 5, pe43.
- Garcia-Mata, R.; Burridge, K. Catching a GEF by its tail. Trends Cell Biol. 2007, 17, 36–43, doi:10.1016/j.tcb.2006.11.004.
- Schiller, M.R. Coupling receptor tyrosine kinases to Rho GTPases—GEFs what’s the link. Cell. Signal. 2006, 18, 1834–1843, doi:10.1016/j.cellsig.2006.01.022.
- Joseph, J.; Radulovich, N.; Wang, T.; Raghavan, V.; Zhu, C.Q.; Tsao, M.S. Rho guanine nucleotide exchange factor ARHGEF10 is a putative tumor suppressor in pancreatic ductal adenocarcinoma. Oncogene 2020, 39, 308–321, doi:10.1038/s41388-019-0985-1.
- Rossman, K.L.; Der, C.J.; Sondek, J. GEF means go: Turning on RHO GTPases with guanine nucleotide-exchange factors. Nat. Rev. Mol. Cell Biol. 2005, 6, 167–180, doi:10.1038/nrm1587.
- Cook, D.R.; Rossman, K.L.; Der, C.J. Rho guanine nucleotide exchange factors: Regulators of Rho GTPase activity in development and disease. Oncogene 2014, 33, 4021–4035, doi:10.1038/onc.2013.362.
- Wu, G.; Li, H.; Yang, Z. Arabidopsis RopGAPs are a novel family of rho GTPase-activating proteins that require the Cdc42/Rac-interactive binding motif for rop-specific GTPase stimulation. Plant Physiol. 2000, 124, 1625–1636.
- Cote, J.F.; Vuori, K. Identification of an evolutionarily conserved superfamily of DOCK180-related proteins with guanine nucleotide exchange activity. J. Cell Sci .2002, 115, 4901–4913, doi:10.1242/jcs.00219.
- Meller, N.; Merlot, S.; Guda, C. CZH proteins: A new family of Rho-GEFs. J. Cell Sci. 2005, 118, 4937–4946, doi:10.1242/jcs.02671.
- DerMardirossian, C.; Bokoch, G.M. GDIs: Central regulatory molecules in Rho GTPase activation. Trends Cell Biol. 2005, 15, 356–363, doi:10.1016/j.tcb.2005.05.001.
- Dransart, E.; Olofsson, B.; Cherfils, J. RhoGDIs revisited: Novel roles in Rho regulation. Traffic 2005, 6, 957–966, doi:10.1111/j.1600-0854.2005.00335.x.
- Ueda, T.; Kikuchi, A.; Ohga, N.; Yamamoto, J.; Takai, Y. Purification and Characterization from Bovine Brain Cytosol of a Novel Regulatory Protein Inhibiting the Dissociation of GDP from and the Subsequent Binding of GTP to rhoB ~20, a ras p214ike GTPbinding Protein. J. Biol. Chem. 1990, 265, 9373–9380.
- Lelias, J.M.; Adra, C.N.; Wulf, G.M.; Guillemot, J.C.; Khagad, M.; Caput, D.; Lim, B. cDNA cloning of a human mRNA preferentially expressed in hematopoietic cells and with homology to a GDP-dissociation inhibitor for the rho GTP-binding proteins. Proc. Natl. Acad. Sci. USA 1993, 90, 1479–1483.
- Scherle, P.; Behrens, T.; Staudt, L.M. Ly-GDI, a GDP-dissociation inhibitor of the RhoA GTP-binding protein, is expressed preferentially in lymphocytes. Proc. Natl. Acad. Sci. USA 1993, 90, 7568–7572.
- Harding, M.A.; Theodorescu, D. RhoGDI2: A new metastasis suppressor gene: Discovery and clinical translation. Urol. Oncol. 2007, 25, 401–406, doi:10.1016/j.urolonc.2007.05.006.
- Abiatari, I.; DeOliveira, T.; Kerkadze, V.; Schwager, C.; Esposito, I.; Giese, N.A.; Huber, P.; Bergman, F.; Abdollahi, A.; Friess, H.; et al. Consensus transcriptome signature of perineural invasion in pancreatic carcinoma. Mol. Cancer Ther. 2009, 8, 1494–1504, doi:10.1158/1535-7163.MCT-08-0755.
- Seraj, M.J.; Harding, M.A.; Gildea, J.J.; Welch, D.R.; Theodorescu, D. The relationship of BRMS1 and RhoGDI2 gene expression to metastatic potential in lineage related human bladder cancer cell lines. Clin. Exp. Metastasis 2001, 18, 519–525.
- Ma, L.; Xu, G.; Sotnikova, A.; Szczepanowski, M.; Giefing, M.; Krause, K.; Krams, M.; Siebert, R.; Jin, J.; Klapper, W. Loss of expression of LyGDI (ARHGDIB), a rho GDP-dissociation inhibitor, in Hodgkin lymphoma. Br. J. Haematol. 2007, 139, 217–223, doi:10.1111/j.1365-2141.2007.06782.x.
- Adra, C.N.; Ko, J.; Leonard, D.; Wirth, L.J.; Cerione, R.A.; Lim, B. Identification of a Novel Protein With GDP Dissociation Inhibitor Activity for the Ras-Like Proteins CDC42Hs and Rac I. Genes Chromosomes Cancer 1993, 8, 253–261.
- Harding, M.A.; Theodorescu, D. RhoGDI signaling provides targets for cancer therapy. Eur. J. Cancer 2010, 46, 1252–1259, doi:10.1016/j.ejca.2010.02.025.
- Xie, F.; Shao, S.; Aziz, A.U.R.; Zhang, B.; Wang, H.; Liu, B. Role of Rho-specific guanine nucleotide dissociation inhibitor alpha regulation in cell migration. Acta Histochem 2017, 119, 183–189, doi:10.1016/j.acthis.2017.01.008.
- Lin, X.; Yang, B.; Liu, W.; Tan, X.; Wu, F.; Hu, P.; Jiang, T.; Bao, Z.; Yuan, J.; Qiang, B.; et al. Interplay between PCBP2 and miRNA modulates ARHGDIA expression and function in glioma migration and invasion. Oncotarget 2016, 7, 19483–19498.
- Gupta, V.; Bhandari, D.; Leyme, A.; Aznar, N.; Midde, K.K.; Lo, I.C.; Ear, J.; Niesman, I.; Lopez-Sanchez, I.; Blanco-Canosa, J.B.; et al. GIV/Girdin activates Galphai and inhibits Galphas via the same motif. Proc. Natl. Acad. Sci. USA 2016, 113, E5721-5730, doi:10.1073/pnas.1609502113.
- Enomoto, A.; Murakami, H.; Asai, N.; Morone, N.; Watanabe, T.; Kawai, K.; Murakumo, Y.; Usukura, J.; Kaibuchi, K.; Takahashi, M. Akt/PKB regulates actin organization and cell motility via Girdin/APE. Dev. Cell 2005, 9, 389–402, doi:10.1016/j.devcel.2005.08.001.
- Le-Niculescu, H.; Niesman, I.; Fischer, T.; DeVries, L.; Farquhar, M.G. Identification and characterization of GIV, a novel Galpha i/s-interacting protein found on COPI, endoplasmic reticulum-Golgi transport vesicles. J. Biol. Chem. 2005, 280, 22012–22020, doi:10.1074/jbc.M501833200.
- Simpson, F.; Martin, S.; Evans, T.M.; Kerr, M.; James, D.E.; Parton, R.G.; Teasdale, R.D.; Wicking, C. A novel hook-related protein family and the characterization of hook-related protein 1. Traffic 2005, 6, 442–458, doi:10.1111/j.1600-0854.2005.00289.x.
- Anai, M.; Shojima, N.; Katagiri, H.; Ogihara, T.; Sakoda, H.; Onishi, Y.; Ono, H.; Fujishiro, M.; Fukushima, Y.; Horike, N.; et al. A novel protein kinase B (PKB)/AKT-binding protein enhances PKB kinase activity and regulates DNA synthesis. J. Biol. Chem. 2005, 280, 18525–18535, doi:10.1074/jbc.M500586200.

