Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Pei Pei Pan | -- | 2956 | 2022-09-29 03:35:27 | | | |
| 2 | Peter Tang | Meta information modification | 2956 | 2022-09-29 06:08:19 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Pan, P.; Huang, X. Molecular Mechanisms of Growth Hormone in Ovarian Functions. Encyclopedia. Available online: https://encyclopedia.pub/entry/27923 (accessed on 21 June 2026).
Pan P, Huang X. Molecular Mechanisms of Growth Hormone in Ovarian Functions. Encyclopedia. Available at: https://encyclopedia.pub/entry/27923. Accessed June 21, 2026.
Pan, Peipei, Xuefeng Huang. "Molecular Mechanisms of Growth Hormone in Ovarian Functions" Encyclopedia, https://encyclopedia.pub/entry/27923 (accessed June 21, 2026).
Pan, P., & Huang, X. (2022, September 29). Molecular Mechanisms of Growth Hormone in Ovarian Functions. In Encyclopedia. https://encyclopedia.pub/entry/27923
Pan, Peipei and Xuefeng Huang. "Molecular Mechanisms of Growth Hormone in Ovarian Functions." Encyclopedia. Web. 29 September, 2022.
Copy Citation
Growth hormone (GH) has been used as a co-gonadotrophin in assisted reproduction, particularly in poor ovarian responders. The application of GH has been alleged to activate primordial follicles and improve oocyte quality, embryo quality, and steroidogenesis. However, the effects of GH on the live birth rate among women is controversial. Additionally, although the basic biological mechanisms that lead to the above clinical differences have been investigated, they are not yet well understood. The actions of GH are mediated by GH receptors (GHRs) or insulin-like growth factors (IGFs). GH regulates the vital signal transduction pathways that are involved in primordial follicular activation, steroidogenesis, and oocyte maturation.
growth hormone
insulin-like growth factor
steroidogenesis
primordial follicle activation
poor ovarian responder
assisted reproductive technology
1. Introduction
Growth hormone (GH) is a 191-amino acid that is generally considered to be secreted by adenohypophysis cells. Adjuvant treatment with GH in assisted reproductive technology (ART) plays a vital role in follicle development, steroidogenesis, oocyte maturation, ovulation, corpus luteum (CL) function, oocyte quality, and ovarian response to the administration of exogenous hormones [1][2][3][4]. GH reaches the target cell membranes, binds to the GH receptors (GHRs), triggers signaling inside cells, and then produces a series of physiological effects. The gene expression of GH is not confined to the pituitary gland, since its mRNA and the protein that is coded by it are produced in many extrapituitary sites, such as testes and ovaries [5][6]. Substantial studies have shown the presence of GH/GHRs in the ovaries of various organisms, including fish, chicken, rats, mice, horses, pigs, monkeys, and humans [7][8][9][10][11][12][13]. The occurrence of GHRs in ovaries indicates that these organs are a target site of GH action. GHRs are expressed in ovarian granulosa cells, theca cells, oocytes, cumulus cells, mammary glands, placentae, and the uterus [14]. In addition, GH and its receptors are expressed in all compartments of ovaries and change according to physiological conditions [15]. Since GH is involved in the regulation of reproductive functions, it has been used as a therapeutic option in ART for more than 30 years, especially in poor ovarian responders (PORs) [16][17][18][19][20][21], polycystic ovary syndrome (PCOS) [22][23], and poor embryonic development [24][25]. However, the effects of GH supplementation on the clinical outcomes, especially live birth rates (LBRs), for those undergoing ART remains controversial, and the underlying mechanisms are still not clear.
2. Molecular Mechanisms of Growth Hormone in Ovarian Functions
In addition to more powerful extensive studies, the molecular mechanisms of GH that are involved in ovarian functions (e.g., ovarian follicular growth [4], the activation of primordial follicles [26][27][28][29], steroidogenesis, oocyte quality, etc.) also need our attention.
2.1. Effects of Growth Hormone on the Regulation of Primordial Follicles
According to the follicle developmental stage and gonadotropin dependence, their development can be classified into the following three phases: gonadotropin-independent phase, gonadotropin-responsive phase, and gonadotropin-dependent phase. After birth, primordial follicles are the major follicles in the ovaries, and then some of them begin to grow into primary follicles. This process is called the activation of primordial follicles, which is associated with the following pathways: (a) the activation of the phosphatidylinositol 3-kinase (PI3K)-phosphatase and tensin homolog (PTEN)-protein kinase B (AKT)-forkhead box O3 (FOX3) signaling pathway [26][27]; (b) the activation of the mammalian target of the rapamycin (mTOR) signaling pathway; and (c) the inhibition of AMH [28][29]. The deregulation of these signaling pathways in oocytes contributes to pathological conditions, including premature ovarian failure (POF) and infertility. Accumulated studies have demonstrated that the effects of GH/IGF on primordial follicle activation could be mediated by the PI3K-PTEN-AKT-FOX3 and mTOR signaling pathways and AMH in GCs and oocytes (Figure 1).
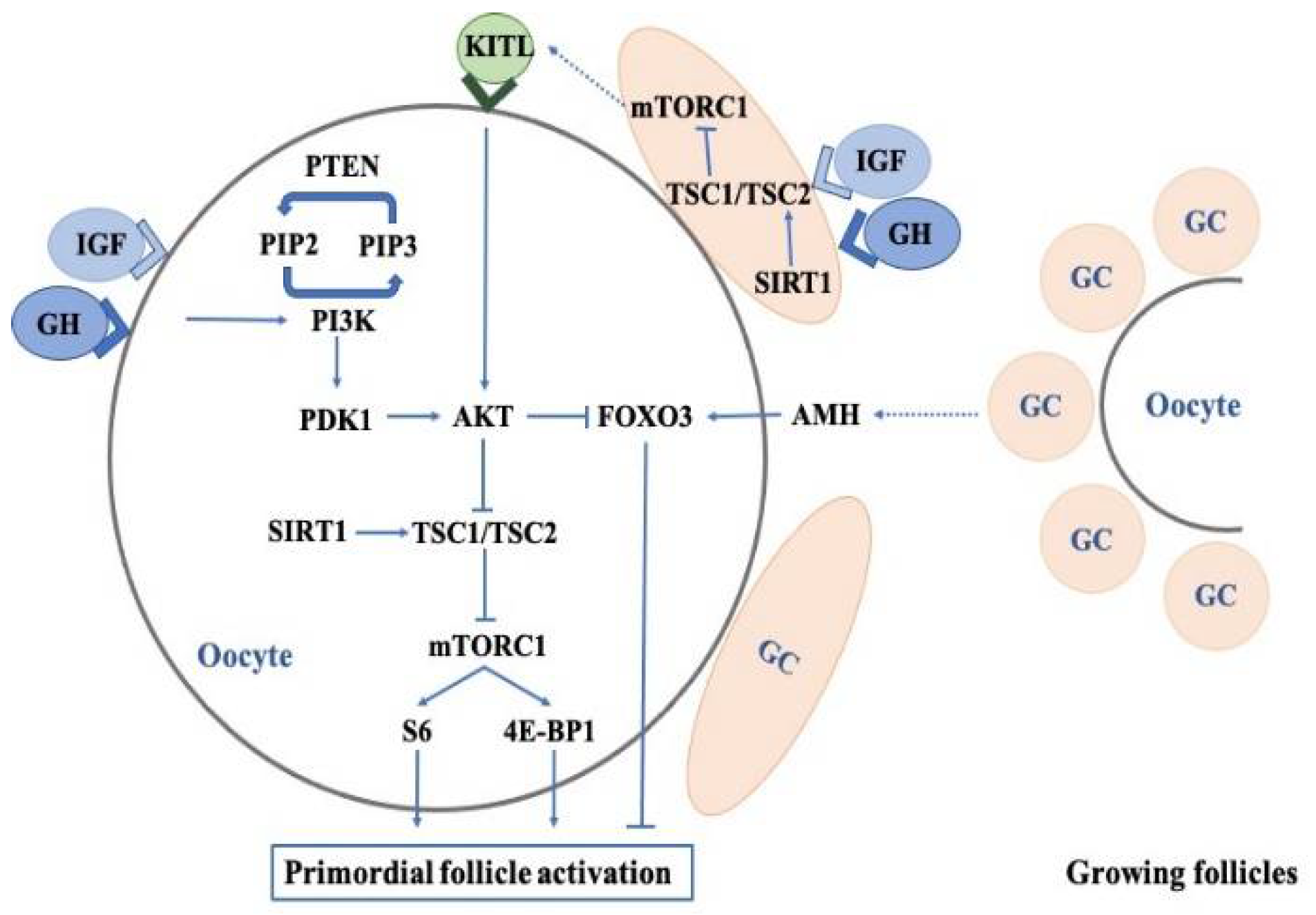
Figure 1. The pathways and their roles in the regulation of primordial follicle activation and quiescence.
Multiple pathways, including PI3K-PTEN-AKT-FOXO3, mTOR-KITL, GH/IGF1, AMH, and SIRT, converge in the mammalian ovaries to regulate the quiescence and activation of primordial follicles.
The abbreviations used are as follows: GH, growth hormone; IGF1, insulin-like growth factor 1; GC, granulosa cell; KITL, KIT ligand; PIP2, phopsphatidylinositol bisphosphate; PIP3, phosphatidylinositol triphosphate; PTEN, phosphatase and tensin homolog; PDK1, phosphoinositide-dependent kinase-1; AKT, protein kinase B; FOXO3, forkhead box O3; PI3K, phosphatidylinositol 3 kinase; AMH, anti-Müllerian hormone; SIRT1, sirtuin 1; TSC1, tuberous sclerosis complex 1; TSC2, tuberin; mTORC1, mTOR complex 1; S6, ribosomal protein S6; 4E-BP1, eukaryotic initiation factor 4E-binding protein 1.
Young female GHR knockout mice have shown increased numbers of primordial follicles and lower serum IGF1, but also harbored a decreased number of follicles in the antral and preovulatory stages [4][30][31], which suggested a slower rate of primordial follicle activation in the above mice. In addition, Saccon et al. also confirmed similar results in GH-deficient Ames Dwarf mice (df/df mice), and further found that df/df mice had fewer GCs surrounding oocytes than normal/df mice in both primordial and primary follicles, and as well as smaller oocyte nuclei diameters in the primordial follicles and oocyte diameters in secondary follicles [32][33]. The oocytes in the primordial and primary follicles of overexpressed bovine GH mice had increased nuclei and oocyte diameters [33]. GH treatment enhanced the progression of primordial follicles into primary follicles, and promoted follicle growth and viability in mouse and goat follicles that were cultured in vitro [34][35].
2.2. Effects of Growth Hormone on Oocyte Quality
Oocyte quality impacts early embryonic survival, as well as the establishment and maintenance of pregnancy and fetal development; thus, it is also regarded as a factor for developmental competence. During folliculogenesis, the oocytes achieve developmental competence. In human ovaries, GHR has been detected in the membranes of cumulus cells [36] and in the nuclei of mature oocytes [13], which confirmed that GH acted at this level to improve the nuclear and cytoplasmic maturation of mature oocytes, and the expansion of cumulus cells as well [12][37][38]. Additionally, high GH concentrations in human follicular fluid has been reported to be associated with the high developmental competence of oocytes [39]. However, the underlying mechanisms of GH that affect oocyte quality are not entirely known.
2.3. Effects of Growth Hormone on the Sensitivity of Oocytes to Gonadotropins
In some studies, GH supplementation has been shown to reduce the need for gonadotropin and shorten the stimulation time, which suggests that supplementary GH could be capable of increasing the sensitivity of oocytes to gonadotropins [40][41][42]. Indeed, GHR/GH-binding protein knockout mice have been reported to have a significantly lower responsiveness to exogenous gonadotropin treatment [4][31]. Additionally, in vivo and ex vivo studies have revealed that GH supplementation as a part of IVF treatment upregulated the expressions of LHR, FSHR, and GHR in human GCs, when isolated after egg collection from women with decreased ovarian reserves [43][44]. FSHR and LHR signaling in GCs is required for follicular selection and dominant follicle formation. Interestingly, GH also acts to support the maturation process of luteinization by increasing LHR density and by reducing FSHR expression prior to ovulation [44]. The stimulation of protein kinase A (PKA) by GH and the prevention of GH-induced effects by PKA blockers have been reported to suggest that both the stimulatory and inhibitory effects of GH on porcine ovarian cells were probably mediated by the cAMP/PKA system. Based on the above evidence, the beneficial effect of GH on gonadotropin responses are probably via the cAMP/PKA pathway. Interestingly, IGF treatment has not been able to improve either fertility or ovarian responsiveness to exogenous gonadotropins [4]; however, the administration of GH has been experimentally shown to enhance the development of small antral follicles into the gonadotropin-dependent phase in vivo [45]. Thus, the effects of GH on the gonadotropin responses via the regulations of FSHR, LHR, and GHR, as well as the activation of the cAMP/PKA pathway, may be independent from IGF1.
2.4. Effects of Growth Hormone on Granulosa Cells and Thecal Cells
2.4.1. Steroidogenesis
During ovarian steroidogenesis, cholesterol is transferred from outer mitochondrial membranes to the inner mitochondrial membranes with the help of an acute regulatory protein (StAR), and is then converted into pregnenolone by CYP11A1, which is a rate-limiting enzyme [46]. Subsequently, CYP17A1 and HSD3B1 sequentially catalyze pregnenolone into androstenedione. Finally, estradiol is generated by the sequential catalyzation of CYP19A1 and HSD17B7 in the GCs [47]. This steroidogenic process can reflect the follicular development and the functions of GCs and theca cells. GH has been demonstrated to affect steroid productions in ovaries, and various intracellular signaling has been shown to be involved in the above process (Figure 2).
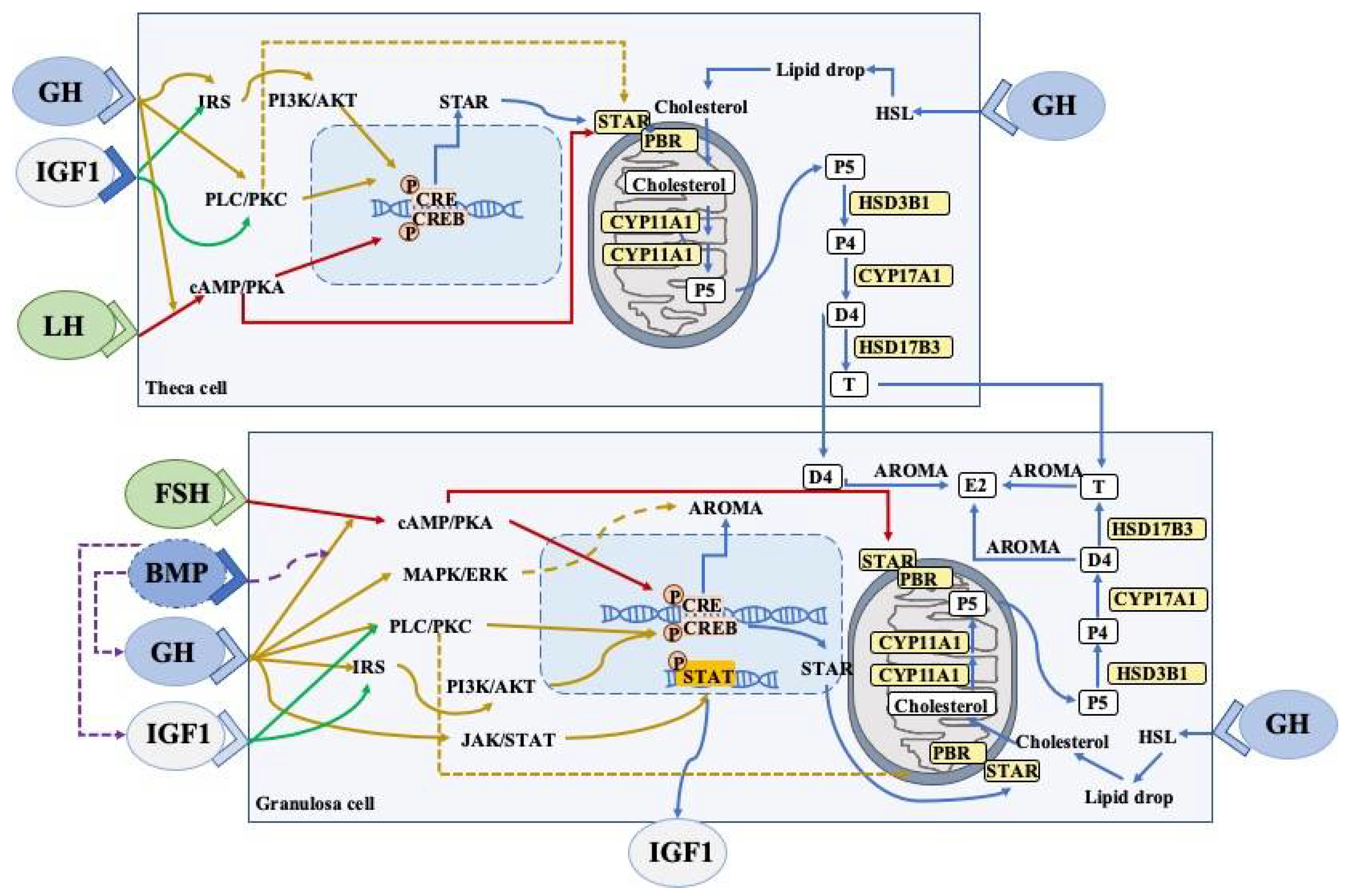
Figure 2. A schematic representation of the major GH/IGF signaling and BMP system networks in ovarian cells (theca and granulosa cells).
GH can induce local IGF1 expression in granulosa cells via JAK-STAT signaling. Both GH and IGF can participate in steroidogenic events and promote cell proliferation via the stimulation of the PLC-PKC and PI3K-AKT pathways, which interact with FSHR and LHR. The expression of aromatase (granulosa cells) and StAR (theca/granulosa cells) is dependent on the CREB pathway. Bone morphogenetic proteins (BMP) suppresses GHR, IGF1, and IGF1R expression, whereas the GH/IGF1 axis downregulates the BMP receptors in the ovaries. Cell proliferation can also be promoted by estrogen and testosterone via autocrine mechanisms.
The abbreviations used are as follows: LHR, luteinising hormone receptor; FSHR, follicle stimulating hormone receptor; cAMP, cyclic AMP; PKA, protein kinase A; CRE, cAMP response element; CREB, cAMP response element binding protein; PLC, phospholipase C, IRS, insulin receptor substrate; PI3K-AKT, phosphoinositide 3-kinase/protein kinase B; StAR, steroidogenic acute regulatory protein; P4, progesterone; P5, pregnenolone; E2, estradiol; D4, androstenedione; T, testosterone; STAT, signal transducer and activator of transcription; AROMA, aromatase, HSL, hormone-sensitive lipase. Arrow, stimulation; Dotted arrows, inhibition.
By binding to the GHRs in the GCs and thecal cells of Graafian follicles, GH has been shown to augment the proliferation of GCs and thecal cells, and to promote steroidogenesis as well [1][48]. Increasing numbers of in vitro studies have found that GH supplementation stimulated the synthesis of progesterone in primary GCs cultures, which could be related to the increased expression of CYP11A1 and HSD3B [8][49]. Moreover, the GH-increased progesterone has been shown to disappear when conditioned media were treated with antiserum against GH [8]. However, there have been contradictory reports about the regulation of GH in the FSH-induced estradiol production [8][49]. Many years ago, 24 normal patients who received short-term GH treatment showed increased E2 production, which was significantly higher than the increase that was induced by FSH injections without GH administration [50]. In isolated preantral caprine follicles, GH has been shown to increase FSH-induced estradiol (E2) production, which could be due to the elevated activity or expression of CYP19A1 [51]. However, another in vitro study found that GH suppressed FSH-induced E2 production, as well as causing reductions in aromatase expression [49]. Interestingly, GH alone has been reported to be unable to induce any steroidogenic responses [50], which implied that the GH steroidogenic effects might be dependent on the FSH actions. Additionally, the incubation of GCs with either recombinant chicken GH or conditioned media that predominantly contained a 15 kDa GH isoform has been shown to significantly increase GC proliferation; meanwhile, the knockdown of local ovarian GH with a specific cGH siRNA in hen GC cultures reduced the proliferation rate of GCs [52]. Thus, GH-mediated disruptions to progesterone and E2 production in GCs may be associated with the expressions of steroidogenic enzymes and the proliferation of GCs.
2.4.2. JAK2-Dependent Signaling Pathway
The binding of GH to the GHR dimer results in the activation of the intracellular Janus Kinase 2 (JAK2) pathway, which can phosphorylate itself and the cytoplasmic regions of GHR [53], and then subsequently phosphorylates STAT molecules (particularly STAT5a, 5b, 1, and 3). STAT5b is considered to be of the utmost importance as it directly regulates the expression of IGF1 [54] and has been shown to act as a mediator of GH-induced IGF1 production in rat GCs [49][55]. When the transcription molecule STAT is recruited, the subsequently mitogen-activated protein kinases (MAPK)/phosphatidylinositol 3-kinase (PI3K)/protein kinase C (PKC)/phospholipase A2 (PLA2) pathway is stimulated. MAPK activation has been demonstrated to regulate FSH-mediated steroidogenesis in the GCs [56][57]. ERK1/2 is involved in the synthesis of progesterone and E2 which is induced by FSH [58]. GH can also enhance FSH-induced ERK signaling, which is likely to be involved in the effects of GH on FSH-induced E2 and progesterone production [49]. Considering that the inhibition of IGF1 blocks the GH-induced stimulation of FSH-induced progesterone production, GH affects the production of steroid hormones via the JAK/STAT and ERK pathways, which are functionally involved in the activation of IGF1 expression. Based on the fact that BMP regulates steroidogenesis and the mitosis of GCs, one study revealed that GH/IGF1 actions impaired BMP-SMA and Mad-related protein 1/5/8 signaling, and that BMP in turn inhibited the IGF1 effects that increased FSH-induced E2 production via the suppression of the expression of the GH/IGF1 system [49]. The effects of GH on the steroidogenesis of GCs have been demonstrated to be enhanced in the presence of the BMP antagonist noggin [49].
2.4.3. JAK2-Independent Signaling Pathway
Other studies have also found that when GH bound to GHR, the PLC/PKC pathway is also activated, which is independent from JAK2 recruitment [55][59]. PKC has been demonstrated to directly trigger cyclic AMP response-element binding protein (CREB)-mediated gene transcription and, subsequently, StAR and/or aromatase expression in GCs, for example [60]. StAR can bind to cholesterol in the cytosol and transport it to outer mitochondrial membranes, where peripheral-type benzodiazepine receptors (PBRs) are involved in its transport from the outer to inner mitochondrial membranes. However, the phosphorylation of StAR by PKC inhibits the transportation of cholesterol in GCs by interfering in StAR-PBR interactions [61].
2.4.4. FSHR Pathway
FSH exerts its actions on GCs mainly through the stimulation of the cAMP-PKA pathway [62][63][64]. CREB, which is a PKA substrate, is known to play an important role in the regulation of the FSH responsive genes (Star and Cyp19a1) in GCs [64][65]. Additionally, the phosphorylation of StAR by PKA stimulates cholesterol transportation by enhancing the interactions between StAR and PBR [61]. Accumulated evidence has shown that GH supplementation could elevate the expressions of LHR, FSHR, and GHR in human GCs [43][44]. Thus, GH can regulate the steroidogenesis of GCs via the FSHR pathway.
2.4.5. GH/IGFs Signaling Pathway
The GH–GHR interaction can induce the production of ovarian IGF1 via the GHR/JAK2/STAT5b pathway [54]. Several GH-deficient mouse models have shown significantly reduced plasma IGF1 concentrations [66][67]. As well as IGF1, other members of the IGF family have also been shown to be influenced by GH [6]. The IGF system comprises two ligands (IGF1 and IGF2), three receptors (IGF1R, IGF2R, and insulin receptors), six secreted IGF-binding proteins (IGFBPs), and IGFBP proteases [68]. Female IGF1R knockout mice have been shown to have ovaries with no antral follicles and demonstrate a 90% reduction in serum E2 levels [69]. Additionally, IGF1 knockout mice have also been shown to exhibit the decreased expression of FSH receptors and, consequently, reduced aromatase expression and E2 secretion [70]. An in vitro study also confirmed that the inhibition of IGF1 signaling restored GH-induced stimulation of FSH-induced progesterone production, which suggested that endogenous IGF1 could be functionally involved with the effects of GH on progesterone production [49]. The biological effects of IGF1 have been further demonstrated to be mediated by IGF1 binding proteins, which are synthesized and secreted in human ovarian GCs [71]. Interestingly, increased free IGF1 has been demonstrated to decrease the synthesis of IGF2R, thereby allowing for more IGF2 to be bioavailable (free) for the induction of steroidogenesis and mitogenesis via the IGF1R [72]. Thus, the above evidence has suggested that by regulating the sensitivity of GCs to gonadotropin, IGF1 and IGF2 could be important downstream factors of GH in female reproduction and that the physiological effects of GH on ovary functions could be due to the direct and/or indirect action of GH.
2.5. Mitochondrial Functions
Oxidative stress is induced by the overproduction of reactive oxygen species (ROS), which are mainly generated by mitochondria [73]. Therefore, mitochondrial dysfunction can lead to pathological changes, disruption to the production of normal ATP levels, and increases in the levels of ROS [74]. Oxidative stress is also considered to be one of the important aspects that causes reproductive dysfunction, including abnormal follicular atresia, ovum meiosis, lower fertilization rates, and delayed embryonic development [75]. Additionally, oxidative stress can initiate during several reproductive diseases, including PCOS [76], endometriosis [77], premature ovarian failure, etc. Mitochondrial dysfunction that is caused by ROS is also involved in reduced oocyte developmental competence and fertilization failure [74]. Recently, GH has been widely applied in the treatment of the above reproductive pathologies, which could be due to its downregulation of oxidative stress in ovaries. GH has been shown to alleviate the total oxidative stress and oxidative stress index level in follicular fluid, and increase GC mitochondrial membrane potential in Chinese patients with PCOS [23]. Additionally, the GCs in the above patients suffered from higher apoptosis, which could be associated with the decreased function of the PI3K-AKT signaling pathway; meanwhile, GH could alleviate caspase-dependent apoptosis of GCs and activate the PI3K-AKT signaling pathway in GCs [78]. Recent studies on mammals have confirmed that the PI3K-AKT signaling pathway could regulate the growth and apoptosis of GCs during follicular development [79][80]. Similarly, GH has been found to reduce ROS-induced apoptosis in some types of cells including the vascular endothelium, cardiomyocytes, and neural and skeletal muscle cells, by activating the PI3K-AKT signaling pathway [81][82]. Furthermore, GH has been shown to exert protective effects on cisplatin-induced ovarian GC apoptosis by downregulating oxidative stress and enhancing mitochondrial functions (i.e., mitochondrial membrane potential and mtDNA copy numbers) via the Sirt3-Sod2 pathway [83]. Sirt3 exists in the mitochondrial matrices and participates in the regulation of mitochondrial functions, as well as acting as an oxidative stress sensor and playing a protective role in controlling ROS generation [84]. In addition to directly reducing the production of ROS, Sirt3 can also regulate the acetylation levels of manganese superoxide dismutase (Sod2), thereby promoting the detoxification of ROS and suppressing mitochondrial oxidative stress [83]. Thus, GH may alleviate apoptosis by lowering the levels of ROS, elevating mitochondrial membrane potential, and recovering mtDNA copy numbers in GCs.
References
- Dosouto, C.; Calaf, J.; Polo, A.; Haahr, T.; Humaidan, P. Growth Hormone and Reproduction: Lessons Learned From Animal Models and Clinical Trials. Front. Endocrinol. 2019, 10, 404.
- Kolibianakis, E.M.; Venetis, C.A.; Diedrich, K.; Tarlatzis, B.C.; Griesinger, G. Addition of growth hormone to gonadotrophins in ovarian stimulation of poor responders treated by in-vitro fertilization: A systematic review and meta-analysis. Hum. Reprod. Update 2009, 15, 613–622.
- Zhang, Y.; Zhang, C.; Shu, J.; Guo, J.; Chang, H.-M.; Leung, P.C.K.; Sheng, J.-Z.; Huang, H. Adjuvant treatment strategies in ovarian stimulation for poor responders undergoing IVF: A systematic review and network meta-analysis. Hum. Reprod. Update 2020, 26, 247–263.
- Bachelot, A.; Monget, P.; Imbert-Bolloré, P.; Coshigano, K.; Kopchick, J.J.; Kelly, P.A.; Binart, N. Growth hormone is required for ovarian follicular growth. Endocrinology 2002, 143, 4104–4112.
- Banerjee, S.; Chaturvedi, C.M. Specific neural phase relation of serotonin and dopamine modulate the testicular activity in Japanese quail. J. Cell. Physiol. 2019, 234, 2866–2879.
- Devesa, J.; Caicedo, D. The Role of Growth Hormone on Ovarian Functioning and Ovarian Angiogenesis. Front. Endocrinol. 2019, 10, 450.
- Kajimura, S.; Kawaguchi, N.; Kaneko, T.; Kawazoe, I.; Hirano, T.; Visitacion, N.; Grau, E.G.; Aida, K. Identification of the growth hormone receptor in an advanced teleost, the tilapia (Oreochromis mossambicus) with special reference to its distinct expression pattern in the ovary. J. Endocrinol. 2004, 181, 65–76.
- Ahumada-Solórzano, S.M.; Carranza, M.E.; Pedernera, E.; Rodríguez-Méndez, A.J.; Luna, M.; Arámburo, C. Local expression and distribution of growth hormone and growth hormone receptor in the chicken ovary: Effects of GH on steroidogenesis in cultured follicular granulosa cells. Gen. Comp. Endocrinol. 2012, 175, 297–310.
- Zhao, J.; Taverne, M.A.M.; van der Weijden, G.C.; Bevers, M.M.; van den Hurk, R. Immunohistochemical localisation of growth hormone (GH), GH receptor (GHR), insulin-like growth factor I (IGF-I) and type I IGF-I receptor, and gene expression of GH and GHR in rat pre-antral follicles. Zygote 2002, 10, 85–94.
- Steffl, M.; Schweiger, M.; Mayer, J.; Amselgruber, W.M. Expression and localization of growth hormone receptor in the oviduct of cyclic and pregnant pigs and mid-implantation conceptuses. Histochem. Cell Biol. 2009, 131, 773–779.
- Marchal, R.; Caillaud, M.; Martoriati, A.; Gérard, N.; Mermillod, P.; Goudet, G. Effect of growth hormone (GH) on in vitro nuclear and cytoplasmic oocyte maturation, cumulus expansion, hyaluronan synthases, and connexins 32 and 43 expression, and GH receptor messenger RNA expression in equine and porcine species. Biol. Reprod. 2003, 69, 1013–1022.
- de Prada, J.K.N.; VandeVoort, C.A. Growth hormone and in vitro maturation of rhesus macaque oocytes and subsequent embryo development. J. Assist. Reprod. Genet. 2008, 25, 145–158.
- Abir, R.; Garor, R.; Felz, C.; Nitke, S.; Krissi, H.; Fisch, B. Growth hormone and its receptor in human ovaries from fetuses and adults. Fertil. Steril. 2008, 90, 1333–1339.
- Xu, Y.-M.; Hao, G.-M.; Gao, B.-L. Application of Growth Hormone in in vitro Fertilization. Front. Endocrinol. 2019, 10, 502.
- Hrabia, A. Growth hormone production and role in the reproductive system of female chicken. Gen. Comp. Endocrinol. 2015, 220, 112–118.
- Owen, E.J.; West, C.; Mason, B.A.; Jacobs, H.S. Co-treatment with growth hormone of sub-optimal responders in IVF-ET. Hum. Reprod. 1991, 6, 524–528.
- Bassiouny, Y.A.; Dakhly, D.M.R.; Bayoumi, Y.A.; Hashish, N.M. Does the addition of growth hormone to the in vitro fertilization/intracytoplasmic sperm injection antagonist protocol improve outcomes in poor responders? A randomized, controlled trial. Fertil. Steril. 2016, 105, 697–702.
- Li, X.-L.; Wang, L.; Lv, F.; Huang, X.-M.; Wang, L.-P.; Pan, Y.; Zhang, X.-M. The influence of different growth hormone addition protocols to poor ovarian responders on clinical outcomes in controlled ovary stimulation cycles: A systematic review and meta-analysis. Medicine 2017, 96, e6443.
- Chu, K.; Pang, W.; Sun, N.; Zhang, Q.; Li, W. Outcomes of poor responders following growth hormone co-treatment with IVF/ICSI mild stimulation protocol: A retrospective cohort study. Arch. Gynecol. Obstet. 2018, 297, 1317–1321.
- Cai, M.-H.; Liang, X.-Y.; Wu, Y.-Q.; Huang, R.; Yang, X. Six-week pretreatment with growth hormone improves clinical outcomes of poor ovarian responders undergoing in vitro fertilization treatment: A self-controlled clinical study. J. Obstet. Gynaecol. Res. 2019, 45, 376–381.
- Choe, S.-A.; Kim, M.J.; Lee, H.J.; Kim, J.; Chang, E.M.; Kim, J.W.; Park, H.M.; Lyu, S.W.; Lee, W.S.; Yoon, T.K.; et al. Increased proportion of mature oocytes with sustained-release growth hormone treatment in poor responders: A prospective randomized controlled study. Arch. Gynecol. Obstet. 2018, 297, 791–796.
- Homburg, R.; Levy, T.; Ben-Rafael, Z. Adjuvant growth hormone for induction of ovulation with gonadotrophin-releasing hormone agonist and gonadotrophins in polycystic ovary syndrome: A randomized, double-blind, placebo controlled trial. Hum. Reprod. 1995, 10, 2550–2553.
- Gong, Y.; Luo, S.; Fan, P.; Jin, S.; Zhu, H.; Deng, T.; Quan, Y.; Huang, W. Growth hormone alleviates oxidative stress and improves oocyte quality in Chinese women with polycystic ovary syndrome: A randomized controlled trial. Sci. Rep. 2020, 10, 18769.
- Li, J.; Chen, Q.; Wang, J.; Huang, G.; Ye, H. Does growth hormone supplementation improve oocyte competence and IVF outcomes in patients with poor embryonic development? A randomized controlled trial. BMC Pregnancy Childbirth 2020, 20, 310.
- Chen, Q.-L.; Shuai, J.; Chen, W.-H.; Zhang, X.-D.; Pei, L.; Huang, G.-N.; Ye, H. Impact of growth hormone supplementation on improving oocyte competence in unexplained poor embryonic development patients of various ages. Gynecol. Endocrinol. Off. J. Int. Soc. Gynecol. Endocrinol. 2021, 38, 1–7.
- Gallardo, T.D.; John, G.B.; Bradshaw, K.; Welt, C.; Reijo-Pera, R.; Vogt, P.H.; Touraine, P.; Bione, S.; Toniolo, D.; Nelson, L.M.; et al. Sequence variation at the human FOXO3 locus: A study of premature ovarian failure and primary amenorrhea. Hum. Reprod. 2008, 23, 216–221.
- Orisaka, M.; Miyazaki, Y.; Shirafuji, A.; Tamamura, C.; Tsuyoshi, H.; Tsang, B.K.; Yoshida, Y. The role of pituitary gonadotropins and intraovarian regulators in follicle development: A mini-review. Reprod. Med. Biol. 2021, 20, 169–175.
- Carlsson, I.B.; Scott, J.E.; Visser, J.A.; Ritvos, O.; Themmen, A.P.N.; Hovatta, O. Anti-Müllerian hormone inhibits initiation of growth of human primordial ovarian follicles in vitro. Hum. Reprod. 2006, 21, 2223–2227.
- Rodgers, R.J.; Abbott, J.A.; Walters, K.A.; Ledger, W.L. Translational Physiology of Anti-Müllerian Hormone: Clinical Applications in Female Fertility Preservation and Cancer Treatment. Front. Endocrinol. 2021, 12, 689532.
- Zaczek, D.; Hammond, J.; Suen, L.; Wandji, S.; Service, D.; Bartke, A.; Chandrashekar, V.; Coschigano, K.; Kopchick, J. Impact of growth hormone resistance on female reproductive function: New insights from growth hormone receptor knockout mice. Biol. Reprod. 2002, 67, 1115–1124.
- Chandrashekar, V.; Zaczek, D.; Bartke, A. The consequences of altered somatotropic system on reproduction. Biol. Reprod. 2004, 71, 17–27.
- Saccon, T.D.; Rovani, M.T.; Garcia, D.N.; Mondadori, R.G.; Cruz, L.A.X.; Barros, C.C.; Bartke, A.; Masternak, M.M.; Schneider, A. Primordial follicle reserve, DNA damage and macrophage infiltration in the ovaries of the long-living Ames dwarf mice. Exp. Gerontol. 2020, 132, 110851.
- Saccon, T.D.; Moreira, F.; Cruz, L.A.; Mondadori, R.G.; Fang, Y.; Barros, C.C.; Spinel, L.; Bartke, A.; Masternak, M.M.; Schneider, A. Ovarian aging and the activation of the primordial follicle reserve in the long-lived Ames dwarf and the short-lived bGH transgenic mice. Mol. Cell. Endocrinol. 2017, 455, 23–32.
- Liu, X.; Andoh, K.; Yokota, H.; Kobayashi, J.; Abe, Y.; Yamada, K.; Mizunuma, H.; Ibuki, Y. Effects of growth hormone, activin, and follistatin on the development of preantral follicle from immature female mice. Endocrinology 1998, 139, 2342–2347.
- Martins, F.S.; Celestino, J.J.H.; Saraiva, M.V.A.; Chaves, R.N.; Rossetto, R.; Silva, C.M.G.; Lima-Verde, I.B.; Lopes, C.A.P.; Campello, C.C.; Figueiredo, J.R. Interaction between growth differentiation factor 9, insulin-like growth factor I and growth hormone on the in vitro development and survival of goat preantral follicles. Braz. J. Med. Biol. Res. 2010, 43, 728–736.
- Ménézo, Y.J.; el Mouatassim, S.; Chavrier, M.; Servy, E.J.; Nicolet, B. Human oocytes and preimplantation embryos express mRNA for growth hormone receptor. Zygote 2003, 11, 293–297.
- Pereira, G.R.; Lorenzo, P.L.; Carneiro, G.F.; Ball, B.A.; Bilodeau-Goeseels, S.; Kastelic, J.; Pegoraro, L.M.C.; Pimentel, C.A.; Esteller-Vico, A.; Illera, J.C.; et al. The involvement of growth hormone in equine oocyte maturation, receptor localization and steroid production by cumulus-oocyte complexes in vitro. Res. Vet. Sci. 2013, 95, 667–674.
- Li, Y.; Liu, H.; Yu, Q.; Liu, H.; Huang, T.; Zhao, S.; Ma, J.; Zhao, H. Growth Hormone Promotes in vitro Maturation of Human Oocytes. Front. Endocrinol. 2019, 10, 485.
- Mendoza, C.; Ruiz-Requena, E.; Ortega, E.; Cremades, N.; Martinez, F.; Bernabeu, R.; Greco, E.; Tesarik, J. Follicular fluid markers of oocyte developmental potential. Hum. Reprod. 2002, 17, 1017–1022.
- Dakhly, D.M.R.; Bassiouny, Y.A.; Bayoumi, Y.A.; Hassan, M.A.; Gouda, H.M.; Hassan, A.A. The addition of growth hormone adjuvant therapy to the long down regulation protocol in poor responders undergoing in vitro fertilization: Randomized control trial. Eur. J. Obstet. Gynecol. Reprod. Biol. 2018, 228, 161–165.
- Kucuk, T.; Kozinoglu, H.; Kaba, A. Growth hormone co-treatment within a GnRH agonist long protocol in patients with poor ovarian response: A prospective, randomized, clinical trial. J. Assist. Reprod. Genet. 2008, 25, 123–127.
- Dogan, S.; Cicek, O.S.Y.; Demir, M.; Yalcinkaya, L.; Sertel, E. The effect of growth hormone adjuvant therapy on assisted reproductive technologies outcomes in patients with diminished ovarian reserve or poor ovarian response. J. Gynecol. Obstet. Hum. Reprod. 2021, 50, 101982.
- Weall, B.M.; Al-Samerria, S.; Conceicao, J.; Yovich, J.L.; Almahbobi, G. A direct action for GH in improvement of oocyte quality in poor-responder patients. Reproduction 2015, 149, 147–154.
- Regan, S.L.P.; Knight, P.G.; Yovich, J.L.; Arfuso, F.; Dharmarajan, A. Growth hormone during in vitro fertilization in older women modulates the density of receptors in granulosa cells, with improved pregnancy outcomes. Fertil. Steril. 2018, 110, 1298–1310.
- Kaiser, G.G.; Kölle, S.; Boie, G.; Sinowatz, F.; Palma, G.A.; Alberio, R.H. In vivo effect of growth hormone on the expression of connexin-43 in bovine ovarian follicles. Mol. Reprod. Dev. 2006, 73, 600–606.
- Payne, A.H.; Hales, D.B. Overview of steroidogenic enzymes in the pathway from cholesterol to active steroid hormones. Endocr. Rev. 2004, 25, 947–970.
- Miller, W.L.; Auchus, R.J. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr. Rev. 2011, 32, 81–151.
- Kobayashi, J.; Mizunuma, H.; Kikuchi, N.; Liu, X.; Andoh, K.; Abe, Y.; Yokota, H.; Yamada, K.; Ibuki, Y.; Hagiwara, H. Morphological assessment of the effect of growth hormone on preantral follicles from 11-day-old mice in an in vitro culture system. Biochem. Biophys. Res. Commun. 2000, 268, 36–41.
- Nakamura, E.; Otsuka, F.; Inagaki, K.; Miyoshi, T.; Matsumoto, Y.; Ogura, K.; Tsukamoto, N.; Takeda, M.; Makino, H. Mutual regulation of growth hormone and bone morphogenetic protein system in steroidogenesis by rat granulosa cells. Endocrinology 2012, 153, 469–480.
- Lanzone, A.; Fortini, A.; Fulghesu, A.M.; Soranna, L.; Caruso, A.; Mancuso, S. Growth hormone enhances estradiol production follicle-stimulating hormone-induced in the early stage of the follicular maturation. Fertil. Steril. 1996, 66, 948–953.
- Ferreira, A.C.A.; Maside, C.; Sá, N.A.R.; Guerreiro, D.D.; Correia, H.H.V.; Leiva-Revilla, J.; Lobo, C.H.; Araújo, V.R.; Apgar, G.A.; Brandão, F.Z.; et al. Balance of insulin and FSH concentrations improves the in vitro development of isolated goat preantral follicles in medium containing GH. Anim. Reprod. Sci. 2016, 165, 1–10.
- Ahumada-Solórzano, S.M.; Martínez-Moreno, C.G.; Carranza, M.; Ávila-Mendoza, J.; Luna-Acosta, J.L.; Harvey, S.; Luna, M.; Arámburo, C. Autocrine/paracrine proliferative effect of ovarian GH and IGF-I in chicken granulosa cell cultures. Gen. Comp. Endocrinol. 2016, 234, 47–56.
- Kopchick, J.J.; Parkinson, C.; Stevens, E.C.; Trainer, P.J. Growth hormone receptor antagonists: Discovery, development, and use in patients with acromegaly. Endocr. Rev. 2002, 23, 623–646.
- Takahashi, Y. The Role of Growth Hormone and Insulin-Like Growth Factor-I in the Liver. Int. J. Mol. Sci. 2017, 18, 1447.
- Dehkhoda, F.; Lee, C.M.M.; Medina, J.; Brooks, A.J. The Growth Hormone Receptor: Mechanism of Receptor Activation, Cell Signaling, and Physiological Aspects. Front. Endocrinol. 2018, 9, 35.
- Kang, S.K.; Tai, C.J.; Cheng, K.W.; Leung, P.C. Gonadotropin-releasing hormone activates mitogen-activated protein kinase in human ovarian and placental cells. Mol. Cell. Endocrinol. 2000, 170, 143–151.
- Seger, R.; Hanoch, T.; Rosenberg, R.; Dantes, A.; Merz, W.E.; Strauss, J.F., 3rd; Amsterdam, A. The ERK signaling cascade inhibits gonadotropin-stimulated steroidogenesis. J. Biol. Chem. 2017, 292, 8847.
- Moore, R.K.; Otsuka, F.; Shimasaki, S. Role of ERK1/2 in the differential synthesis of progesterone and estradiol by granulosa cells. Biochem. Biophys. Res. Commun. 2001, 289, 796–800.
- Rowlinson, S.W.; Yoshizato, H.; Barclay, J.L.; Brooks, A.J.; Behncken, S.N.; Kerr, L.M.; Millard, K.; Palethorpe, K.; Nielsen, K.; Clyde-Smith, J.; et al. An agonist-induced conformational change in the growth hormone receptor determines the choice of signalling pathway. Nat. Cell Biol. 2008, 10, 740–747.
- Manna, P.R.; Huhtaniemi, I.T.; Stocco, D.M. Mechanisms of protein kinase C signaling in the modulation of 3’,5’-cyclic adenosine monophosphate-mediated steroidogenesis in mouse gonadal cells. Endocrinology 2009, 150, 3308–3317.
- Niswender, G.D. Molecular control of luteal secretion of progesterone. Reproduction 2002, 123, 333–339.
- Casarini, L.; Crépieux, P. Molecular Mechanisms of Action of FSH. Front. Endocrinol. 2019, 10, 305.
- Puri, P.; Little-Ihrig, L.; Chandran, U.; Law, N.C.; Hunzicker-Dunn, M.; Zeleznik, A.J. Protein Kinase A: A Master Kinase of Granulosa Cell Differentiation. Sci. Rep. 2016, 6, 28132.
- Liang, A.; Plewes, M.R.; Hua, G.; Hou, X.; Blum, H.R.; Przygrodzka, E.; George, J.W.; Clark, K.L.; Bousfield, G.R.; Butnev, V.Y.; et al. Bioactivity of recombinant hFSH glycosylation variants in primary cultures of porcine granulosa cells. Mol. Cell. Endocrinol. 2020, 514, 110911.
- Mukherjee, A.; Park-Sarge, O.K.; Mayo, K.E. Gonadotropins induce rapid phosphorylation of the 3’,5’-cyclic adenosine monophosphate response element binding protein in ovarian granulosa cells. Endocrinology 1996, 137, 3234–3245.
- Słuczanowska-Głąbowska, S.; Laszczyńska, M.; Piotrowska, K.; Głąbowski, W.; Kopchick, J.J.; Bartke, A.; Kucia, M.; Ratajczak, M.Z. Morphology of ovaries in laron dwarf mice, with low circulating plasma levels of insulin-like growth factor-1 (IGF-1), and in bovine GH-transgenic mice, with high circulating plasma levels of IGF-1. J. Ovarian Res. 2012, 5, 18.
- Isola, J.V.V.; Zanini, B.M.; Sidhom, S.; Kopchick, J.J.; Bartke, A.; Masternak, M.M.; Stout, M.B.; Schneider, A. 17α-Estradiol promotes ovarian aging in growth hormone receptor knockout mice, but not wild-type littermates. Exp. Gerontol. 2020, 129, 110769.
- Jones, J.I.; Clemmons, D.R. Insulin-like growth factors and their binding proteins: Biological actions. Endocr. Rev. 1995, 16, 3–34.
- Baumgarten, S.C.; Armouti, M.; Ko, C.; Stocco, C. IGF1R Expression in Ovarian Granulosa Cells Is Essential for Steroidogenesis, Follicle Survival, and Fertility in Female Mice. Endocrinology 2017, 158, 2309–2318.
- Zhou, J.; Kumar, T.R.; Matzuk, M.M.; Bondy, C. Insulin-like growth factor I regulates gonadotropin responsiveness in the murine ovary. Mol. Endocrinol. 1997, 11, 1924–1933.
- Lavranos, T.C.; O’Leary, P.C.; Rodgers, R.J. Effects of insulin-like growth factors and binding protein 1 on bovine granulosa cell division in anchorage-independent culture. J. Reprod. Fertil. 1996, 107, 221–228.
- Spicer, L.J.; Aad, P.Y. Insulin-like growth factor (IGF) 2 stimulates steroidogenesis and mitosis of bovine granulosa cells through the IGF1 receptor: Role of follicle-stimulating hormone and IGF2 receptor. Biol. Reprod. 2007, 77, 18–27.
- Das, M.; Sauceda, C.; Webster, N.J.G. Mitochondrial Dysfunction in Obesity and Reproduction. Endocrinology 2021, 162, bqaa158.
- Roth, Z. Symposium review: Reduction in oocyte developmental competence by stress is associated with alterations in mitochondrial function. J. Dairy Sci. 2018, 101, 3642–3654.
- Wang, L.; Tang, J.; Wang, L.; Tan, F.; Song, H.; Zhou, J.; Li, F. Oxidative stress in oocyte aging and female reproduction. J. Cell. Physiol. 2021, 236, 7966–7983.
- Ostadmohammadi, V.; Jamilian, M.; Bahmani, F.; Asemi, Z. Vitamin D and probiotic co-supplementation affects mental health, hormonal, inflammatory and oxidative stress parameters in women with polycystic ovary syndrome. J. Ovarian Res. 2019, 12, 5.
- Samimi, M.; Pourhanifeh, M.H.; Mehdizadehkashi, A.; Eftekhar, T.; Asemi, Z. The role of inflammation, oxidative stress, angiogenesis, and apoptosis in the pathophysiology of endometriosis: Basic science and new insights based on gene expression. J. Cell. Physiol. 2019, 234, 19384–19392.
- Gong, Y.; Luo, S.; Fan, P.; Zhu, H.; Li, Y.; Huang, W. Growth hormone activates PI3K/Akt signaling and inhibits ROS accumulation and apoptosis in granulosa cells of patients with polycystic ovary syndrome. Reprod. Biol. Endocrinol. 2020, 18, 121.
- John, G.B.; Shidler, M.J.; Besmer, P.; Castrillon, D.H. Kit signaling via PI3K promotes ovarian follicle maturation but is dispensable for primordial follicle activation. Dev. Biol. 2009, 331, 292–299.
- Grosbois, J.; Demeestere, I. Dynamics of PI3K and Hippo signaling pathways during in vitro human follicle activation. Hum. Reprod. 2018, 33, 1705–1714.
- Caicedo, D.; Díaz, O.; Devesa, P.; Devesa, J. Growth Hormone (GH) and Cardiovascular System. Int. J. Mol. Sci. 2018, 19, 290.
- Chung, J.-Y.; Kim, H.-J.; Kim, M. The protective effect of growth hormone on Cu/Zn superoxide dismutase-mutant motor neurons. BMC Neurosci. 2015, 16, 1.
- Wang, J.; Wu, J.; Zhang, Y.; Zhang, J.; Xu, W.; Wu, C.; Zhou, P. Growth hormone protects against ovarian granulosa cell apoptosis: Alleviation oxidative stress and enhancement mitochondrial function. Reprod. Biol. 2021, 21, 100504.
- Zhang, T.; Zhou, Y.; Li, L.; Wang, H.-H.; Ma, X.-S.; Qian, W.-P.; Shen, W.; Schatten, H.; Sun, Q.-Y. SIRT1, 2, 3 protect mouse oocytes from postovulatory aging. Aging 2016, 8, 685–696.
More
Information
Subjects:
Obstetrics & Gynaecology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
974
Revisions:
2 times
(View History)
Update Date:
29 Sep 2022
Table of Contents




Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No