Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Costel Moldoveanu | -- | 3908 | 2022-09-28 06:42:30 | | | |
| 2 | Jason Zhu | Meta information modification | 3908 | 2022-09-29 03:46:29 | | | | |
| 3 | Jason Zhu | -1 word(s) | 3907 | 2022-09-30 07:40:07 | | | | |
| 4 | Jason Zhu | Meta information modification | 3907 | 2022-10-09 03:28:44 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Zbancioc, G.; Mangalagiu, I.I.; Moldoveanu, C. Synthesis of Fluorescent Five- and Six-Membered Ring Azaheterocycles. Encyclopedia. Available online: https://encyclopedia.pub/entry/27757 (accessed on 22 July 2026).
Zbancioc G, Mangalagiu II, Moldoveanu C. Synthesis of Fluorescent Five- and Six-Membered Ring Azaheterocycles. Encyclopedia. Available at: https://encyclopedia.pub/entry/27757. Accessed July 22, 2026.
Zbancioc, Gheorghita, Ionel I. Mangalagiu, Costel Moldoveanu. "Synthesis of Fluorescent Five- and Six-Membered Ring Azaheterocycles" Encyclopedia, https://encyclopedia.pub/entry/27757 (accessed July 22, 2026).
Zbancioc, G., Mangalagiu, I.I., & Moldoveanu, C. (2022, September 28). Synthesis of Fluorescent Five- and Six-Membered Ring Azaheterocycles. In Encyclopedia. https://encyclopedia.pub/entry/27757
Zbancioc, Gheorghita, et al. "Synthesis of Fluorescent Five- and Six-Membered Ring Azaheterocycles." Encyclopedia. Web. 28 September, 2022.
Copy Citation
The synthesis of fluorescent azaheterocycles continues to arouse strong interest due to their great potential for application as sensors and biosensors, luminophores on in the construction of Organic Light-Emitting Diode (OLED) devices, laser and other semiconductor devices, as well as to their potential biological properties as antimicrobial, antifungal, anticancer, antituberculosis antioxidant and anti-HIV agents. The advantages of the azaheterocyclic fluorophores, such as small size, enriched photostability, a wide and tunable spectral range, and, frequently, high brightness, are the reason why these fluorophores are preferred and used in various medical application. Probe structure can be modified to adjust excitation and emission wavelengths, target-binding affinity, chemical reactivity, and subcellular localization.
five and six membered ring azaheterocycles
fluorescence
optical properties
1. Five Membered Ring Azaheterocycles
Five membered ring azaheterocycles, such as pyrroles, diazoles, triazoles, tetrazoles and their fused derivatives, are privileged scaffolds posing a wide range of biological activities and diverse applications in organic electronics [1][2][3][4][5][6][7][8][9]. Due to these properties, many researchers were focused on the obtaining of new compounds with such skeleton.
Martínez-Lara et al. [10] developed a new obtaining methodology of two different angular indolocarbazole moieties by using two sequential gold- and molybdenum-catalysis to obtain new indolo [2,3-c]carbazoles as potential luminophores with application in OLED devices.
A selection of 1,1-bis(indol-3-yl)-3-alkyn-2-ols 3, was readily prepared through the alkylation of the indole derivatives 1 with methyl or phenyl glyoxal, followed by a standard Sonogashira-type reaction with 2-nitroaryl iodides. Next, these compounds underwent a cyclization reaction, promoted by NaAuCl4, affording the corresponding carbazoles 4 (see Scheme 1).
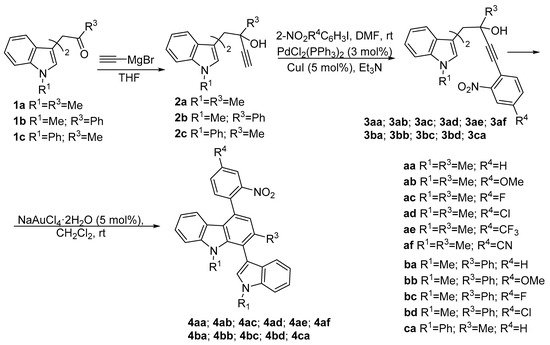
Scheme 1. Synthetic procedure used for the obtaining of indolylcarbazole derivatives 4.
Without further purification, the crude carbazoles were directly subjected to Mo-catalyzed Cadogan reaction delivering the desired indolocarbazoles 5. All indolocarbazoles were obtained in high yields. The researchers [10] used microwave irradiation for the synthesis of the indolocarbazole derivatives 4 and 5, and obtained final products in much shorter time and with better yields (see Scheme 2).
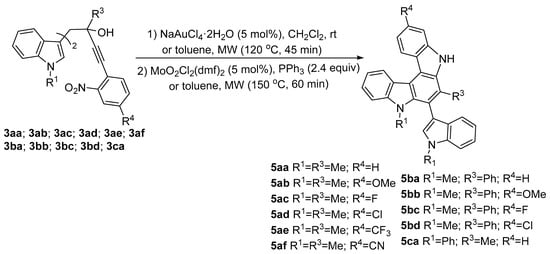
Scheme 2. Synthesis of indolo[2,3-c]carbazole derivatives.
The phenyl-substituted 5ba indolo[2,3-c]carbazole shows a wider absorption wavelength range than methylsubstituted 5aa indolo[2,3-c]carbazole and both show fluorescence quantum yields around 70% in DMSO, double as the Φfl of some similar compounds from the literature. These compounds are potentially useful for optoelectronic applications due to their high extinction coefficient and fluorescence quantum yields.
By combining 2H-imiazole 1-oxides 7a–l with pentafluorophenyl lithium 8 made in situ from pentafluorobenzene 6 and n-BuLi, Moseev et al. [11] were able to create novel pentafluoroaryl-modified derivatives of 2H-imidazole type 10a–l and 11a–l, as is presented in Scheme 3. Pentafluorophenyllithium 8 first attacks the imidazole-N-oxide 7a–l, resulting in the unstable σH-adduct 9 that can be converted into the product either by “addition-elimination” (Path A), leading to the formation of compounds 10a–l, or by “addition–oxidation” (Path B), leading to the creation of compounds 11a–l containing N-oxide group.
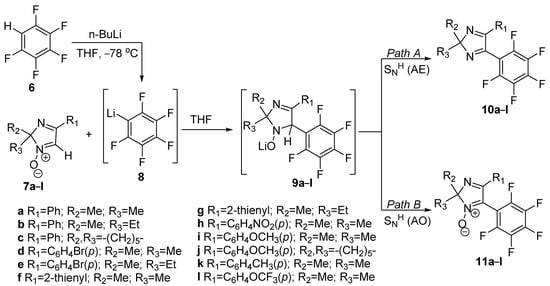
Scheme 3. Synthesis of pentafluoroaryl-modified derivatives.
As possible applications, the researchers claim that the push-pull fluorophore systems containing both electron-donating (EDG) and electron-withdrawing (EWG) groups in the vicinal position could be used as push–pull fluorophore systems. The obtained molecule types 10 and 11 have undergone a photophysical study to determine the potential practical uses in the construction of photoactive materials.
The researchers concluded that the presence of EWG in the para-position of the phenyl group improved the fluorescence quantum yield of the produced fluorophores by measuring the quantum yield of polyfluorinated-2Himidazole derivatives 10 and 11.
The phenomenon of intramolecular charge transfer (ICT) in polar protonic solvents (MeOH and EtOH) has been shown for several compounds, and connections between “structure-photophysical characteristics” have been found. The characteristics of the investigated photophysical properties allow them to adjust the structures of the photoactive molecules, which opens the possibility of using the synthesized push-pull systems in the development of difficult fluorometric molecular sensors.
Pu et al. [12] synthesized antipyrine-containing diarylethenes derivatives 14 in their attempt to obtain novel multi-controllable photochromic and fluorescent derivatives (see Scheme 4).
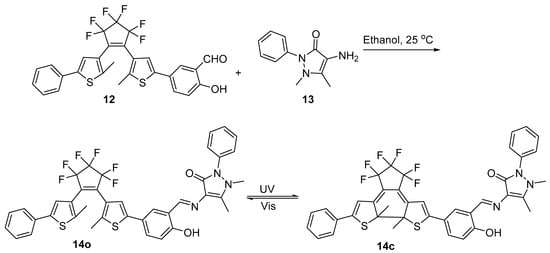
Scheme 4. Synthesis of antipyrine-containing diarylethenes derivatives 14.
It was determined that 14o had an absolute fluorescence quantum yield of 1.4%. UV light (297 nm) irradiation caused the photocyclization reaction, which resulted in the synthesis of 14c with a quantum yield of 0.8%. As a result, the emission strength of 14o abruptly reduced, and the fluorescence changed from bright orange to darkness. Conversely, by exposing 14o with appropriate visible light, the fluorescence may be recovered.
At 580 nm, the isomer 14o showed high brightly orange fluorescence. The emission strength of 14o reduced to 18% with the addition of 5.0 equiv of tetrabutylammonium hydroxide, and the color changed noticeably from bright orange to blackness as a result of the creation of deprotonated 14o’. The fluorescence could return to its initial form upon neutralization with 2.0 equiv of HCl. Similar to this, the stimulation of TBAH/HCl in acetonitrile could reversibly alter the fluorescence of 14c. When irradiated to UV/vis light, the fluorescence alterations between 14o’ and 14c’ was irreversible. The fluorescence of 14o’ stayed unaltered when irradiated to UV light, however when exposed to visible light (λ > 500 nm), the fluorescence of 14c’ might return to that of 14o’, as may be observed in Scheme 5. The results obtained by the researchers shown that the base could suppress the photochromism of diarylethene with an antipyrine unit’s and acid could restore it.
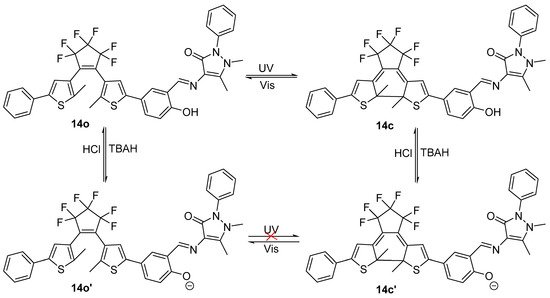
Scheme 5. Structure changes of 14 induced by TBAH/HCl and UV/vis.
In order to examine the fluorescence variations of 14o, 5.0 equiv of several metal ions, including Al3+, Zn2+, Ni2+, K+, Cd2+, Ca2+, Cu2+, Ba2+, Pb2+, Cr3+, Co2+, Sr2+, Mg2+, Mn2+, Hg2+ and Fe3+, were added. The researchers s pointed out that the addition of Al3+ was the only metal ion that significantly affected the fluorescence of 14o; other metal ions had a negligible effect. As a result of this, the diarylethene with an antipyrine unit type 14o demonstrated special base-gated photochromic properties and could be used as a highly selective naked-eye chemosensor for Al3+ detection.
2. Six Membered Ring Azaheterocycles
Electron-deficient six membered ring azaheterocycles, such as azines and diazines, are strongly desired and studied due to their potential biological activities and also to their potential applications in organic electronics [13][14][15][16][17].
Motivated by the small size and high quantum yield (Φ = 60%) of the unsubstituted pyridin-2-amine, being a potential scaffold for a fluorescent probe, Li et al. [18], in their research for new multisubstituted aminopyridines, used the Rh-catalyzed coupling of vinyl azide with isonitrile to form a vinyl carbodiimide intermediate, which followed a tandem cyclization with an alkyne to the desired amine.
The reactions were carried “one-pot”, using vinyl azide 15 and isonitrile 16, Rh-catalyst, proper ligand and 1,4-dioxane as solvent, under N2 atmosphere and at room temperature in the first stage, then after the vinyl azide spot disappeared on TLC, NH4Cl, NaHCO3, and alkyne were added, and the mixture was heated to 120 °C for 8 h (see Scheme 6).
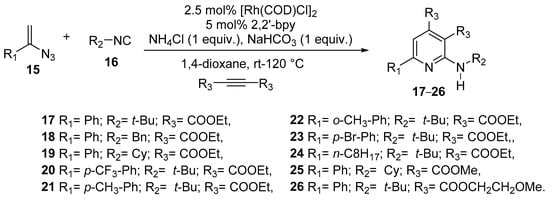
Scheme 6. One-pot synthesis of multi-substituted aminopyridines 17–26.
Compound 17 was derivatized in order to observe the variation of the photoluminescent properties, as is presented in Scheme 7.
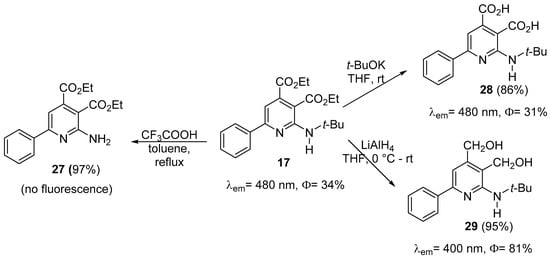
Scheme 7. Derivatization of multisubstituted aminopyridine 17.
Thus, when the tertiary butyl group was cleaved using CF3COOH the obtained compound 27 showed no fluorescence. When the ester groups were hydrolyzed to carboxylic acid, the obtained compound 28 still had a good fluorescence (Φ = 31%). When compound 35 was reduced with LiAlH4 a high fluorescent (Φ = 81%) alcohol 29 was obtained in excellent yield, but the λem was reduced to 400 nm.
The same researchers [18], in order to apply the newly synthesized compound as fluorescent probe, used the click reaction to bind the fluorescent aminopyridine scaffold to biomolecules conjugated with alkynyl group (see Scheme 8). Firstly, they synthesized a Boc-NH protected compound 30 accordingly to the above-described method from corresponding starting materials, then the azido substituted derivative 31 was obtained via a Sandmeyer reaction. Finally, the azide 31 react easily with phenylacetylene or propynol (by using catalytic amounts of Cu(II)) to obtain the desired triazole products 32 or 33.
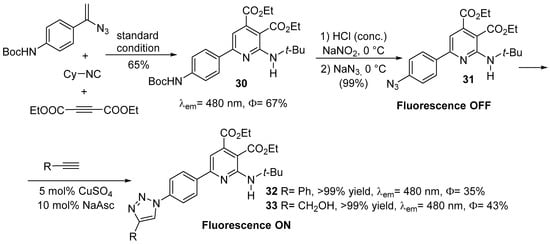
Scheme 8. Binding the fluorescent aminopyridine scaffold to biomolecules conjugated with alkynyl group.
The researchers successfully applied this “click-and-probing” experiment to bovine serum albumin (BSA) conjugated with a terminal alkyne, demonstrating the biochemical application of this probe and of the fluorescent multisubstituted aminopyridines for detection and analysis.
Piloto et al. [19] studied the use of acridine azaheterocycle as a photochemically removable protecting group for the carboxylic group of amino acids involved in neurotransmision. In this respect, a series of amino acids 34 (glycine, alanine, glutamic acid, β-alanine and γ-aminobutyric acid) protected on amino group with tert-butoxycarbonyl, was treated with 9-bromomethylacridine 35, the corresponding heterocyclic ester 36 being obtained (see Scheme 9). The coupling reaction requires potassium fluoride as base and N,N-dimethylformamide as solvent, and take place at room temperature.
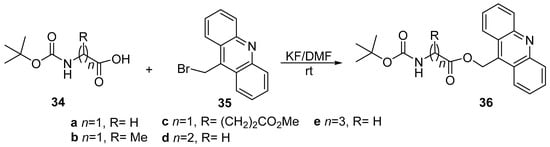
Scheme 9. The reaction of tert-butoxycarbonyl amino protected amino acids with 9-bromomethylacridine 35.
Due to the heterocyclic chromophore, the absorption maximum of compounds showed no influence of amino acid residue. Ester derivatives 36a–e displayed emission maxima between 411–435 nm in both solvents, with moderate Stokes’ shifts (50–75 nm). These esters exhibited higher fluorescence quantum yields in a methanol–HEPES buffer solvent system.
The deprotection of the carboxylic group from the corresponding fluorescent derivatives in methanol-HEPES buffer (80:20) solutions was achieved by irradiation. The best results on photorelease of these amino acids from were obtained on irradiation at 350 nm. This ability of acridinyl methyl ester make them a suitable option for the photochemical release of functional molecules bearing a carboxylic acid group in organic synthesis and in cellular applications.
In order to obtain new luminogens for sensing strong acids Tang et al. [20] used Suzsuki cross-coupling reaction. The luminogens consist of one pyridine, 1,3-diazine, 1,4-diazine, 1,2-diazine and phthalazine moieties as the central cores and two AIE-active tetraphenylethene units in the lateral sides. Aggregation induced emission (AIE) effect exhibiting high emission in concentrated solution or even in the solid state, hold great promise for a luminogen in practical applications.
PY-TPE 39b, PYM-TPE 39c, PYA-TPE 39d, PYD-TPE 39e and PTZ-TPE 39f were easily synthesized by 4-(1,2,2-triphenylvinyl) phenyl boronic acid 37 reaction with 2,5-dibromopyridine 38b, 2,5-dibromopyrimidine 38c, 2,5-dibromopyrazine 38d, 3,6-dibromopyridazine 38e, 1,4-dibromophthalazine 38f under Suzuki cross-coupling in high yields (see
Scheme 10). For comparison, an analogue molecule BZ-TPE 38a fused with phenyl unit was also prepared.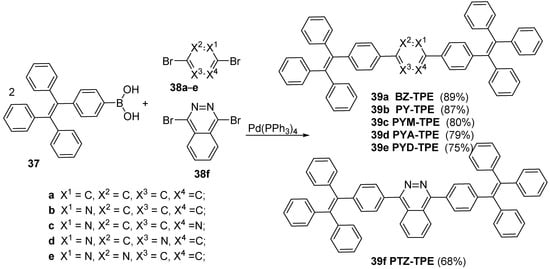
Scheme 10. Suzuki cross-coupling reaction of 4-(1,2,2-triphenylvinyl) phenyl boronic acid 37 with 2,5-dibromopyridine 38b, 2,5-dibromopyrimidine 38c, 2,5-dibromopyrazine 38d, 3,6-dibromopyridazine 38e, 1,4-dibromophthalazine 38f.
The emission spectra (in CH3CN–water) were recorded in order to determine the AIE effect in aggregate states of the as-prepared luminogens. Benzyl-, pyridine-, pyrimidine- and pyrazine-tetraphenylethenes (39a–d) exhibited high fluorescence quantum yields in aggregate states due to a typical AIE effect. 1,2-Diazine-tetraphenylethenes (pyridazine-39e and phthalazine-39f) have strong yellow fluorescence emission under protonation enabling AIE performance. The synergetic effect of AIE and the protonation on the 1, 2-diazine segments allows them to detect strong acids with very low pKa values.
The similarities of the 4-hydroxy-1,3-thiazole unit, a chromophore and fluorophore, with naturally occurring luciferin, determine Menzel et al. [21] to obtain new of arylamine-modified thiazoles as donor-acceptor dyes. The donor part of the dyes was based on the 4-methoxy-1,3-thiazole core substituted with an arylamine (phenyl-, p-anisole-, p-tolyl-, or phenothiazine-) in the 5-position as donor, and a pyridine, pyrimidine, or pyrazine moiety in the 2-position as acceptor.
The synthesis of the 4-hydroxy-1,3-thiazoles 42a–c involved a Hantzsch thiazole cyclizations between the corresponding azaheterocyclic thioamides 40a–c and ethyl 2-bromo-2-(4-nitrophenyl)acetate 41a. Similarly, starting from ethyl 2-bromo-2-(4-bromophenyl)acetate 41b and pyridine-2-carbothioamide 40a, compound 42d was prepared, then methylated via Williamson ether synthesis, with methyl iodide in DMSO. The reduction of the nitro group in 43a–c with freshly prepared Raney nickel and hydrazine in EtOH fallowed (see Scheme 11).
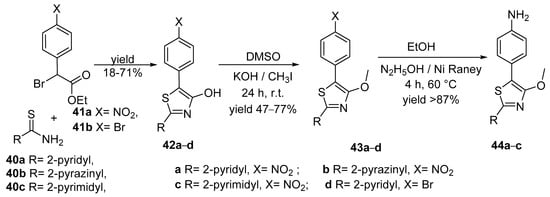
Scheme 11. Synthesis of new arylamine-modified thiazoles donor-acceptor dyes.
The reaction condition for the double N-arylation Buchwald–Hartwig cross-coupling reaction were: bis(dibenzylideneacetone)palladium(0) [Pd(dba)2] as the precatalyst, KOtBu as the base, and toluene as solvent. By using the P(tBu)3 reagent, the desired products were obtained in moderate to good yields (76–90%). The obtaining of both disubstituted A1 and B1, and the monosubstituted products A1m (69%) and B1m (88%), demonstrates the two-step nature of the reaction (see Scheme 12).
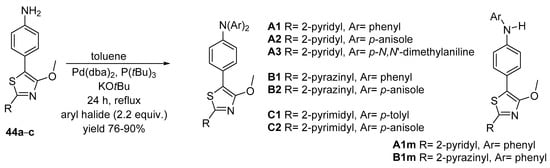
Scheme 12. N-arylation Buchwald–Hartwig cross-coupling reaction of arylamines 44a–c.
The Buchwald–Hartwig reaction starting with 43d, a thiazole substituted aryl halide, was not successful under the conditions of the double N-arylation of the amines 44a–c, the biarylphosphane 2-(dicyclohexylphosphanyl)-2′,6′-dimethoxy-1,1′-biphenyl (SPHOS) was required as ligand (see Scheme 13).
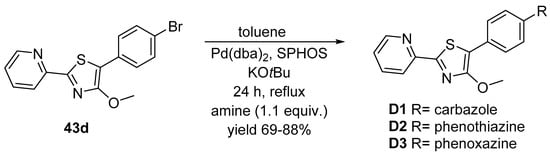
Scheme 13. N-arylation Buchwald–Hartwig cross-coupling reaction of thiazole substituted aryl halide 43d.
The arylamine donors has a strong influence on the emission quantum yields recorded in CH3CN solution at room temp, the phenyl-based triarylamines having good quantum yields (40–47%), while the p-anisole-based triarylamines show values below 1% for quantum yields, and consequently no fluorescence for the p-N,N′-dimethylaniline derivative. The obtained photoluminescence quantum yields are correlated with the measured emission lifetimes (τ).
Only the emission due to the carbazole moiety, as a main band, can be observed for compound D1 in the polar CH3CN, while for D2 and D3 the emissions are modest.
The researchers [21] carried out measurements of the absorptions (and emissions) in different solvents for compounds A2 and D1 in order to investigate them in more detail. The increasing of the solvent polarity induces a weak hypsochromic shift on the absorption maxima, but a bathochromic shift on the emission maxima. This behavior is due to a conformational transformation from a planar locally excited (LE) state to an intramolecular charge-transfer (ICT) or, consequently, to a twisted intramolecular charge-transfer (TICT) state.
Menzel et al. [21] tested also the ability of these dyes to act as ligands and synthesized seven new ruthenium complexes and measured their emission spectra. The synthesis of the heteroleptic RuII complexes involve the activation of the cis-(dmbpy)2RuCl2 precursor with AgPF6 prior to the complexation in acetone with the appropriate ligand (1 equiv.) for 24 h under reflux conditions (see Scheme 14).
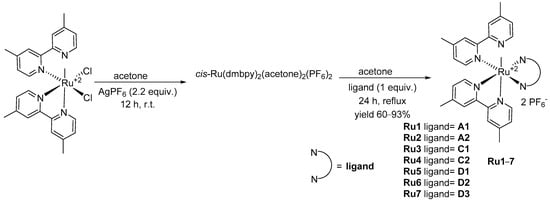
Scheme 14. The synthesis of the heteroleptic RuII complexes.
Each complex shows an enhanced absorption in the visible part of the UV/Vis spectrum, due to additional ligand-centered (LC) π–π* transitions and metal-to-ligand charge-transfer (MLCT) transitions originating from 4-methoxy-1,3-thiazole ligands.
3. Fused Azaheterocycles
Fused azaheterocycles combine an electron-excessive pyrrole and an electron deficient azine ring which involve an uneven π-electron distribution. This uneven π-electron distribution induces interesting optical properties on the compound with such fused azaheterocycles and make them very attractive materials in optoelectronics [22][23]. Moreover, the variety of potential applications in the fields of medicinal of these compounds explains the increased interest of researchers in their study [24][25][26].
Rajbongshi et al. [22] obtained new chromeno[2,3-b]indoles 47a–c as part of the series of chromenoindole derivatives by reacting 1-(α-amino-α-arylalkyl)-2-naphthol 45 with indole 46 at 100 °C in the presence of catalytic p-toluenesulfonic acid, I2 and tert-butyl hydroperoxide (TBHP), as is presented in Scheme 15.
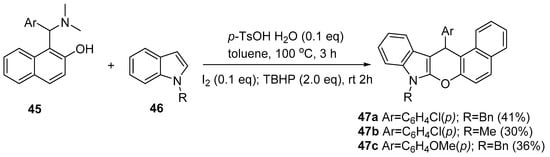
Scheme 15. Synthesis of chromeno[2,3-b]indoles 47a–c.
In various solvents, the fluorescence spectra of the produced chromeno[2,3-b]indoles 47a–c alter and show a significant Stokes shift, ranging from 250 nm in acetonitrile to 186 nm in ethyl acetate. The chromeno[2,3-b]indoles’ fluorescence quantum yield was evaluated at 280 nm using tryptophan in water as a reference and at 313 nm using naphthalene.
The investigated chromeno[2,3-b]indoles 47a–c produce fluorescence at a significantly lower energy compared to their absorption, and their fluorescence spectra exhibit a solvatochromic shift, which suggests intramolecular charge transfer in the excited state. The three chromeno[2,3-b]indoles’ with strong Stokes-shifted fluorescence suggests that they may be used as luminescent solar concentrators or scintillators.
Li et al. [23] obtained a fused five-membered azaheterocycle with an aggregation-induced emission (AIE) characteristic using an unanticipated regioselective photoreaction.
The starting compound o-TPBQ 48 were easily prepared through a facile one-step synthetic route via a modified Sonogashira coupling reaction according to the previously reported literature [27][28].
The researchers proposed all of the potential products, including the common six-membered ones, 50 C6-TPBQ′ and 51 C6-TPBQ″, as well as the five-membered ring product 49 (C5-TPBQ), although the likelihood of this is very low. They also considered the specific location of the N atom in o-TPBQ and the reported literature on the photoreaction. Surprisingly, the typical six-membered cyclized product (C6-TPBQ’ or C6-TPBQ”) was not obtained; instead, only the five-membered cyclized product 49, C5-TPBQ, was obtained (see Scheme 16). NMR and high-resolution mass spectroscopies were used to confirm the structure of C5-TPBQ.
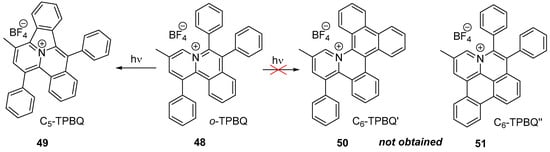
Scheme 16. Regioselective photoreaction of o-TPBQ 48.
In DMSO/water solutions with various water fractions, the photoluminescence spectra of C5-TPBQ were recorded. This compound has a low fluorescence quantum yield in DMSO solution (1.1%), and when water is gradually added to the solution, an increased photoluminescence signal is obtained. The AIE characteristic was demonstrated by the 110-fold increase in emission intensity in DMSO/water mixes with 99% water compared with DMSO solution.
This provides a method for quickly and easily creating fused five-membered azaheterocyclic compounds with unique fluorescence properties. These compounds have a wide range of uses in the biological and optoelectronic domains.
Having in view the synthesis of new 1H-pyrazolo[3,4-b]quinoxaline (PQX) derivatives with specific photophysical properties as potential materials for optoelectronics, Wojtasik et al. [29] tried to improve the Cadogan synthesis of these compounds. The researchers used triphenylphosphine as reducing agent, in dimethylacetamide (DMAc) at 240 °C, under microwave irradiation and obtained PQX derivatives in much shorter time and with better yields of the final product (see Scheme 17).
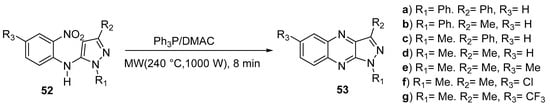
Scheme 17. Cadogan synthesis of 1H-pyrazolo[3,4-b]quinoxaline derivatives 53a–g.
The substrates 52a–g were obtained by the coupling of the appropriate 2-iodonitrobenzene derivatives 54 with 1,3-disubstituted 5-aminopyrrazole 55 in the presence of palladium catalyst and BINAP (2,2′-bis(diphenylphosphino)-1,1′-binaphtyl), as is presented in Scheme 18. By this reaction, researchers obtained the best yields, despite the long reaction time.

Scheme 18. The coupling of 2-iodonitrobenzene derivatives 54 with 1,3-disubstituted 5-aminopyrrazole 55.
The introduction of the electron donor substituent (methyl group-53e) in the 6th position of 1,3-dimethyl-1H-pyrazolo[3,4-b]quinoxaline 53d did not changed the emission properties comparing with the unsubstituted 53d system, while the electron-accepting substituents (chlorine-53f and trifluoromethyl group-53g) shifted the fluorescence band twoards longer wavelenghts by 8 nm to 21 nm, respectively, and induced an increase of the quantum yield of the fluorescence.
In order to develop a new family of thermally activated delayed fluorescence (TADF) emitters, utilizing the donor-acceptor scaffold, Goya et al. [30] focused on the use of the electron-deficient azaaromatic scaffold in place of the usual dibenzo[a,j]phenazine (DBPHZ) unit. Organic compounds that can display TDAF are very intense studied as emitters for efficient OLEDs, since they can achieve theoretically 100% internal quantum efficiency (IQE) by harvesting electrically generated triplet excitation and convert into the emissive singlet excitations through reverse intersystem crossing (rISC).
Pyrido[2,3-b]pyrazine (PYPZ) moiety was selected as the electron-acceptor (A) unit, due to the both pyridine and pyrazine π-deficient heterocycles. When this electron deficient core is connected with an appropriate electron-donor (D), intramolecular charge-transfer (ICT) in the excited state will occur and CT states should become the first singlet excited state (S1). As donor part was selected dihydrophenazasiline (DHPHAzSi) since DHPHAzSi compounds are moderate electron donor.
The 7-bromo-2,3-diphenylpyrido[2,3-b]pyrazine 59 and 10-bromo-acenaphtho[1,2-b]pyrido[2,3-e]pyrazine 61 donors were bonded to the corresponding DHPHAzSi acceptors through the Pd-catalyzed Buchwald-Hartwig amination in good yields (see Scheme 19).
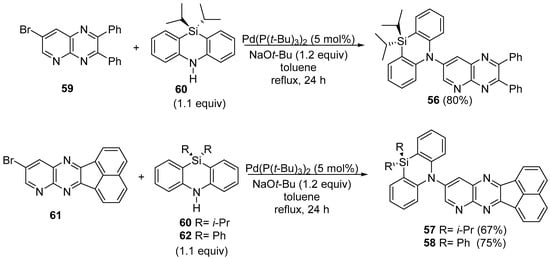
Scheme 19. Buchwald-Hartwig amination of 7-bromo-2,3-diphenylpyrido[2,3-b]pyrazine 59 and 10-bromo-acenaphtho[1,2-b]pyrido[2,3-e]pyrazine 61 donors with DHPHAzSi acceptors.
The starting compounds 59 and 61 were prepared according to the literature procedures [31][32], through the condensation between 2,3-diamino-5-bromopyridine with benzyl and phenanthrene-9,10-dione, respectively.
Compounds 56–58 manifest emissions in two different time domains. The first component is the prompt fluorescence (PF) from the singlet excited state (S1), with a lifetime in the nanosecond time delay, and the second components are the delayed emissions from the high triplet excited state (T2) (as theoretical calculations revealed), decaying in micro to millisecond delay time.
Georgescu et al. [33][34][35] studied the synthesis and fluorescence of 3-aryl-7-benzoyl-pyrrolo[1,2-c]pyrimidines in order to determinate the influence of the chemical structure and of solvent polarity on their optical properties.
The researchers used, for the synthesis of 3-biphenyl-pyrrolo[1,2-c]pyrimidines, a one-pot, three-component procedure [33][34], which involve a 1,3-dipolar cycloaddition reaction of a 4-biphenyl pyrimidinium-N-ylide with an activated alkyne 65 in 1,2-epoxybutane at reflux. The ylide is generated “in situ” from the corresponding pyrimidinium salts that was formed by the N-alkylation of the pyrimidine 63 with halogenoketone 64. The advantage of performing the reaction in a one-pot three-component approach is the direct formation of the final aromatic compounds 66a–j, avoiding the formation of dipyrimidino-pyrazinic inactivated products (see Scheme 20).
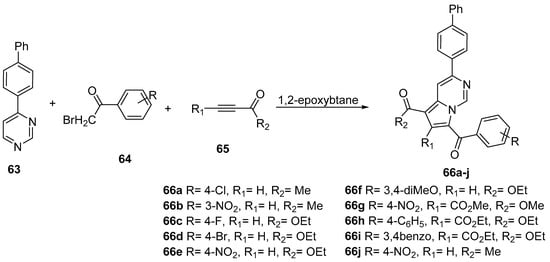
Scheme 20. One-pot, three-component synthesis of 3-biphenyl-pyrrolo[1,2-c]pyrimidines 66a–j.
The absorption and emission spectra of compounds 66a–j were recorded in acetonitrile:chloroform (1:1) (3.5 * 10−6 mol/L) solutions.
In order to calculate the quantum yield (Φfl), according to the Equation (1) researchers need to determine the maximum value of the absorbance at the emission wavelength λem2, (A), area of the emission peak (I), and refractive index (n) for the solution of investigated compound, and quantum yield (Φref), maximum value of the absorbance at the emission wavelength λem2 (Aref), area of the emission peak (Iref), and refractive index (nref) for the standard solution (quinine sulphate), respectively.
It can be seen that two of the compounds (66c p-flouoro substituted and 66f 3,4-dimethoxy substituted) have higher values of quantum yield. These higher values of quantum yield can be explained by a more extended π electron conjugated system in the case of this compound. Ethyl 3-(4-biphenylyl)-7-(3,4-dimethoxybenzoyl)pyrrolo[1,2-c]pyrimidine-5-carboxylate was found to have the highest quantum yield value (55%). These higher quantum yield values suggest that the studied compounds are promising candidates for fluorescent chemical sensors.
In view of future exploitation of pyrrolo[1,2-c]pyrimidine compounds in the field of bio-imaging investigations Georgescu et al. [35] synthesized a series of new pyrrolo[1,2-c]pyrimidine derivatives and studied their photophysical properties. The synthetic approach was similar with the one described above, but this time did not use the one pot strategy, but only by the 1,3-dipolar cycloaddition of the pyrimidinium ylides generated “in situ” from the corresponding pyrimidinium bromides 67 with the alkyne dipolarophiles 68 in 1,2-epoxybutane as reaction medium and acid scavenger (see Scheme 21).
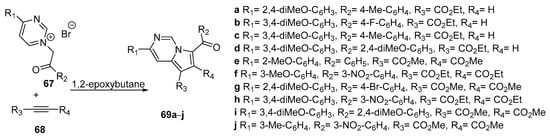
Scheme 21. Synthesis of pyrrolo[1,2-c]pyrimidine 69a–j by 1,3-dipolar cycloaddition.
The photophysical properties of fused derivatives 69a–j were investigated by using different sorts of solvents. In chloroform, compounds derived from the ethyl propiolate, 69a–d, present better fluorescence yield than the compounds derived from the symmetric alkynes 69e–j. The excitation, during the recording of the emission spectra, was performed at λabs2 at which a higher intensity of the fluorescence was obtained. A main emission band is located in the blue region of the visible spectrum in 437–463 nm region.
The researchers [35] investigate the influence of all R1, R2, R3 and R4 substituents on the fluorescence properties of the synthesized compounds and concluded that a substituent that determine the growth of conjugation inside the pyrrolo[1,2-c]pyrimidine fragment lead to an increase of the fluorescence, while a substituent that determine the decrease of conjugation inside the pyrrolo[1,2-c]pyrimidine fragment lead to decrease of the fluorescence.
Tomashenko et al. [36] obtained new pyrido[2,1-a]pyrrolo[3,2-c]isoquinoline 76a–c heterocyclic system by using Pd to catalyze the intramolecular cyclization of 1-[1-benzyl-2-(2-bromophenyl)-1H-pyrrol-3-yl]pyridin-1-ium bromides 74a–c (see Scheme 22).
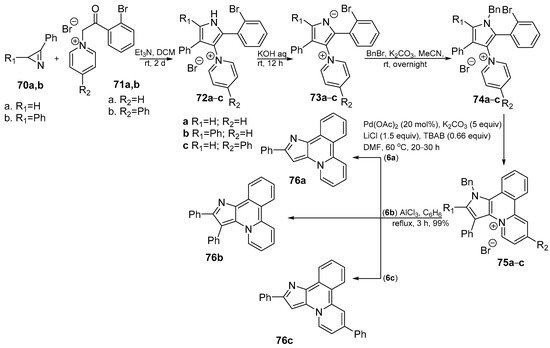
Scheme 22. Pd catalyzed synthesis of pyrido[2,1-a]pyrrolo[3,2-c]isoquinolines 76a–c.
The obtained compounds 76a–c have fluorescent properties in solutions, with quantum yields for 76c in methanol reaching 81%. Photophysical data were studied in toluene, acetonitrile, dichloromethane (DCM) and methanol, respectively. The obtained compounds display moderate to strong fluorescence in solution, giving minor variations in emission maxima position with changes in solvent polarity. The biggest effect was shown in methanol solutions and upon addition of proton donors to aprotic solvents.
References
- Al Matarneh, C.M.; Shova, S.; Mangalagiu, I.I.; Danac, R. Synthesis, structure, antimycobacterial and anticancer evaluation of new pyrrolo-(phenanthroline) derivatives. J. Enzym. Inhib. Med. Chem. 2016, 31, 470–480.
- Thanikachalam, P.V.; Maurya, R.K.; Garg, V.; Monga, V. An insight into the medicinal perspective of synthetic analogs of indole: A review. Eur. J. Med. Chem. 2019, 180, 562–612.
- Danac, R.; Al Matarneh, C.M.; Shova, S.; Daniloaia, T.; Balan, M.; Mangalagiu, I.I. New indolizines with phenanthroline skeleton: Synthesis, structure, antimycobacterial and anticancer evaluation. Bioorg. Med. Chem. 2015, 23, 2318–2327.
- Naim, M.J.; Alam, O.; Nawaz, F.; Alam, M.J.; Alam, P. Current status of pyrazole and its biological activities. J. Pharm. Bioallied Sci. 2016, 8, 2–17.
- Ali, I.; Lone, M.N.; Aboul-Enein, H.Y. Imidazoles as potential anticancer agents. MedChemComm 2017, 8, 1742–1773.
- Mantu, D.; Antoci, V.; Moldoveanu, C.; Zbancioc, G.; Mangalagiu, I.I. Hybrid imidazole (benzimidazole)/pyridine (quinoline) derivatives and evaluation of theiranticancer and antimycobacterialactivity. J. Enzym. Inhib. Med. Chem. 2016, 31, 96–103.
- Bansal, Y.; Silakari, O. The therapeutic journey of benzimidazoles: A review. Bioorg. Med. Chem. 2012, 20, 6208–6236.
- Bozorov, K.; Zhao, J.; Aisa, H.A. 1,2,3-Triazole-containing hybrids as leads in medicinal chemistry: A recent overview. Bioorg. Med. Chem. 2019, 27, 3511–3531.
- Kaushik, N.; Kumar, N.; Kumar, A.; Singh, U.K. Tetrazoles: Synthesis and biological activity. Immunol. Endocr. Metab. Agents Med. Chem. 2018, 18, 3–21.
- Martínez-Lara, F.; Suárez, A.; Suárez-Pantiga, S.; Tapia, M.J.; Sanz, R. Straight access to highly fluorescent angular indolocarbazoles via merging Au- and Mo-catalysis. Org. Chem. Front. J. 2020, 7, 1869–1877.
- Moseev, T.D.; Varaksin, M.V.; Gorlov, D.A.; Charushin, V.N.; Chupakhin, O.N. Transition-Metal-Free C–H/C–Li Coupling of Nonaromatic 2H-Imidazole 1-Oxides with Pentafluorophenyl Lithium in the Design of Novel Fluorophores with Intramolecular Charge Transfer Effect. J. Org. Chem. 2020, 85, 11124–11133.
- Pu, S.; Zhang, C.; Fan, C.; Liu, G. Multi-controllable properties of an antipyrine-based diarylethene and its high selectivity for recognition of Al3+. Dyes Pigments 2016, 129, 24–33.
- Gonçalves, T.; Sameiro, M. Fluorescent labeling of biomolecules with organic probes. Chem. Rev. 2009, 109, 190–212.
- Grammel, M.; Hang, H.C. Chemical reporters for biological discovery. Nat. Chem. Biol. 2013, 9, 475–484.
- Lang, K.; Chin, J.W. Cellular incorporation of unnatural amino acids and bioorthogonal labeling of proteins. Chem. Rev. 2014, 114, 4764–4806.
- Zhang, Y.; Li, S.; Zhang, H.; Xu, H. Design and application of receptor-targeted fluorescent probes based on small molecular fluorescent Dyes. Bioconj. Chem. 2021, 32, 4–24.
- Algar, W.R.; Massey, M.; Rees, K.; Higgins, R.; Krause, K.D.; Darwish, G.H.; Peveler, W.J.; Xiao, Z.; Tsai, H.-Y.; Gupta, R.; et al. Photoluminescent nanoparticles for chemical and biological analysis and imaging. Chem. Rev. 2021, 121, 9243–9358.
- Li, Z.; Li, Y.; Chang, W.; Pang, S.; Li, X.; Duan, L.; Zhang, Z. Synthesis and Fluorescent Properties of Aminopyridines and the Application in “Click and Probing”. Molecules 2022, 27, 1596.
- Piloto, A.M.; Hungerford, G.; Costa, S.P.G.; Gonçalves, M.S.T. Acridinyl methyl esters as photoactive precursors in the release of neurotransmitteramino acids. Photochem. Photobiol. Sci. 2013, 12, 339–347.
- Tang, R.; Wang, X.; Zhang, W.; Zhuang, X.; Bi, S.; Zhanga, W.; Zhang, F. Aromatic azaheterocycle-cored luminogens with tunable physical properties via nitrogen atoms for sensing strong acids. J. Mater. Chem. C 2016, 4, 7640–7648.
- Menzel, R.; Kupfer, S.; Mede, R.; Weiß, D.; Görls, H.; González, L.; Beckert, R. Arylamine-Modified Thiazoles as Donor–Acceptor Dyes: Quantum Chemical Evaluation of the Charge-Transfer Process and Testing as Ligands in Ruthenium(II) Complexes. Eur. J. Org. Chem. 2012, 27, 5231–5247.
- Rajbongshi, B.K.; Bhattacharyya, H.P.; Choitanya; Pegu, D.; Sharma, S.; Baruah, P.K.; Sarma, M. Ultra-High Stokes Shift in Polycyclic ChromenoIndoles. Polycycl. Aromat. Compd. 2022, 42, 1710–1727.
- Li, Q.; Gong, J.; Li, Y.; Zhang, R.; Wang, H.; Zhang, J.; Yan, H.; Lam, J.W.Y.; Sung, H.H.Y.; Williams, I.D.; et al. Unusual light-driven amplification through unexpected regioselective photogeneration of five-membered azaheterocyclic AIEgen. Chem. Sci. 2021, 12, 709–717.
- Zbancioc, A.M.; Miron, A.; Tuchilus, C.; Rotinberg, P.; Mihai, C.T.; Mangalagiu, I.; Zbancioc, G. Synthesis and in vitro analysis of novel dihydroxyacetophenone derivatives with antimicrobial and antitumor activities. Med. Chem. 2014, 10, 476–483.
- Antoci, V.; Oniciuc, L.; Amariucai-Mantu, D.; Moldoveanu, C.; Mangalagiu, V.; Amarandei, A.M.; Lungu, C.N.; Dunca, S.; Mangalagiu, I.I.; Zbancioc, G. Benzoquinoline Derivatives: A Straightforward and Efficient Route to Antibacterial and Antifungal Agents. Pharmaceuticals 2021, 14, 335.
- Popovici, L.; Amarandi, R.M.; Mangalagiu, I.I.; Mangalagiu, V.; Danac, R. Synthesis, molecular modelling and anticancer evaluation of new pyrrolopyridazine and pyrrolophthalazinederivatives. J. Enzym. Inhib. Med. Chem. 2019, 34, 230–243.
- Li, Q.; Li, Y.; Min, T.; Gong, J.; Du, L.; Phillips, D.L.; Liu, J.; Lam, J.W.Y.; Sung, H.H.Y.; Williams, I.D.; et al. Time-Dependent Photodynamic Therapy for Multiple Targets: A Highly Efficient AIE-Active Photosensitizer for Selective Bacterial Elimination and Cancer Cell Ablation. Angew. Chem. Int. Ed. 2020, 59, 9470–9477.
- Han, Y.R.; Shim, S.-H.; Kim, D.-S.; Jun, C.-H. Synthesis of Benzoquinolizinium Salts by Rh(III)-Catalyzed Cascade Double N-Annulation Reactions of Allylamines, Diarylacetylenes, and HBF4. Org. Lett. 2017, 19, 2941–2944.
- Wojtasik, K.; Danel, A.; Wojtasik, M.; Lukasiewicz, M. Synthesis of 1H-Pyrazoloquinoxaline Derivatives by Modification of the Regiospecific Reaction–the Influence of the Microwave Field. ChemistrySelect 2021, 6, 4330–4335.
- Goya, T.; Zimmermann Crocomo, P.; Hosono, T.; Minakata, S.; de Sousa, L.E.; de Silva, P.; Data, P.; Takeda, Y. A New Entry to Purely Organic Thermally Activated Delayed Fluorescence Emitters Based on Pyridopyrazine-Dihydrophenazasilines Donor-Acceptor Dyad. Asian J. Org. Chem. 2022, 11, e202100780.
- Yin, L.; Liebscher, J. Palladium-Catalyzed Cross-Coupling Reactions of 7-Bromo-2,3-diphenylpyridopyrazine. Synthesis 2005, 8, 1345–1349.
- Kaur, G.; Singh, A.; Kaur, N.; Banerjee, B. A general method for the synthesis of structurally diverse quinoxalines and pyrido-pyrazine derivatives using camphor sulfonic acid as an efficient organo-catalyst at room temperature. Synth. Commun. 2021, 51, 1121–1131.
- Tatu, L.; Georgescu, E.; Boscornea, C.; Ungureanu, E.M. Synthesis and fluorescence of 1-pyrimidin-5-yl]ethanone. UPB Bull. Sci. 2015, 77, 49–58.
- Tatu, M.L.; Georgescu, E.; Boscornea, C.; Popa, M.M. Ungureanu, E.M. Synthesis and fluorescence of new 3-biphenylpyrrolopyrimidines. Arab. J. Chem. 2017, 10, 643–652.
- Ungureanu, E.M.; Tatu, M.L.; Georgescu, E.; Boscornea, C.; Popa, M.M.; Stanciu, G. Influence of the chemical structure and solvent polarity on the fluorescence of 3-aryl-7-benzoyl-pyrrolo pyrimidines. Dyes Pigments 2020, 174, 108023.
- Tomashenko, O.A.; Khlebnikov, A.F.; Mosiagin, I.P.; Novikov, M.S.; Grachova, E.V.; Shakirova, J.R.; Tunik, S.P. A new heterocyclic skeleton with highly tunable absorption/emission wavelength via H-bonding. RSC Adv. 2015, 5, 94551–94561.
More
Information
Subjects:
Chemistry, Organic
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.3K
Entry Collection:
Organic Synthesis
Revisions:
4 times
(View History)
Update Date:
09 Oct 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No