Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | fusheng zhang | -- | 4205 | 2022-09-27 08:32:42 | | | |
| 2 | Sirius Huang | Meta information modification | 4205 | 2022-09-28 03:50:54 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Zhang, F.; Cheng, W. Mechanism of Antibiotics Resistance. Encyclopedia. Available online: https://encyclopedia.pub/entry/27634 (accessed on 01 August 2026).
Zhang F, Cheng W. Mechanism of Antibiotics Resistance. Encyclopedia. Available at: https://encyclopedia.pub/entry/27634. Accessed August 01, 2026.
Zhang, Fusheng, Wei Cheng. "Mechanism of Antibiotics Resistance" Encyclopedia, https://encyclopedia.pub/entry/27634 (accessed August 01, 2026).
Zhang, F., & Cheng, W. (2022, September 27). Mechanism of Antibiotics Resistance. In Encyclopedia. https://encyclopedia.pub/entry/27634
Zhang, Fusheng and Wei Cheng. "Mechanism of Antibiotics Resistance." Encyclopedia. Web. 27 September, 2022.
Copy Citation
Bacterial drug resistance is rapidly developing as one of the greatest threats to human health. Bacteria will adopt corresponding strategies to crack the inhibitory effect of antibiotics according to the antibacterial mechanism of antibiotics, involving the mutation of drug target, secreting hydrolase, and discharging antibiotics out of cells through an efflux pump, etc. Bacteria are found to constantly evolve new resistance mechanisms to antibiotics, including target protective protein, changes in cell morphology, and so on, endowing them with multiple defense systems against antibiotics, leading to the emergence of multi-drug resistant (MDR) bacteria and the unavailability of drugs in clinics.
bacterial drug resistance
antibacterial compounds
1. Introduction
Antibiotic resistance is recognized as one of the most serious global threats to human health in the 21st century [1][2][3]. In 2019, researchers found that 1.27 million people died directly from antibiotic resistance according to analyses of cases related to antibiotic resistance in 204 countries and regions around the world, and another 4.95 million people died associated to antibiotic resistance, most of which died from methicillin-resistant Staphylococcus aureus (MRSA), reaching over 100,000 [4][5].
Since Penicillium was demonstrated to inhibit the growth of other bacteria in 1928 [6][7], the antibiotic industry has rapidly developed and successfully saved the lives of thousands of wounded patients, and as such, is regarded as one of the greatest discoveries in human history. In the following decades, plenty of different types of antibiotics were identified and successively applied to clinical treatment. However, a few years after the discovery of Penicillin, the phenomenon of bacterial resistance began to appear. In 1972, MRSA was found in England, the United States, and other countries [8]. In 2008, a metallo-β-lactamase gene blaNDM-1 with the capability to resist the most widely antibacterial hydrocarbon antibiotics was identified for the first time in Klebsiella pneumoniae [9]. In 2015, a new drug-resistance gene mcr-1 was identified in Enterobacteriaceae of pigs in southern China, with the capability to express drug resistance to polymyxins [10]. In 2017, the WHO published its first-ever list of the deadliest superbugs that threaten human health, covering 12 families of dangerous bacteria that have developed resistance to antibiotics, where the “critical” section refers to three bacteria—carbapenem resistant Acinetobacter baumannii, carbapenem resistant Pseudomonas aeruginosa, and carbapenem resistant and ESBL-producing Enterobacteriaceae (including Klebsiella, E. coli, Serratia, and Proteus)—which are all resistant to multiple drugs, and can elicit a range of serious infections [11].
In a natural environment, bacteria are supposed to constantly compete for survival resources, which equips numerous microorganisms with the evolved chemical substances produced in the process of metabolism that can inhibit or kill other microorganisms [12][13], where the Penicillins are metabolites of Penicillium, and cephalosporins are metabolites of Cephalosporium, etc. Under the pressure of survival, competitors have also evolved corresponding resistance mechanisms to various antibiotics, and antibiotic-secreting strains often have corresponding resistance mechanisms to protect themselves. Studies have demonstrated the resistance of archaea from 30,000 years ago to β-lactam antibiotics (e.g., penicillin) and aminoglycoside antibiotics (e.g., streptomycin) [14][15]. It is obvious that bacterial drug resistance is developed as a self-protection mechanism that bacteria retain in natural selection, and various resistance mechanisms of bacteria have been developed against antibiotics by long-term evolution, enabling bacteria to escape the action of more antibiotics, which aggravates the problem of bacterial drug resistance.
Up to now, antibiotics have been widely utilized for more than 80 years globally, and there exist thousands of available antibiotics, with hundreds of them commonly applied in clinical practice. However, since the 1990s, the identification of antibiotics has gradually ground to a halt, and most novel antibiotics are optimized and upgraded only on the basis of the original antibiotics, without changes in the drug targets and antibacterial mechanism. In recent years, with the development of bioinformatics, synthetic biology, and other biotechnology, new potential antibiotics have been increasingly discovered, such as Teixobactin (2015) [16], Chimeric peptidomimetic (2017) [17], Arylomycin (2018) [18], Corbomycin (2020) [19], Iboxamycin (IBX) (2021) [20] and so on, the most of which have new drug targets in comparison to previous antibiotics, with the nonspecific targets enabling a broad antibacterial spectrum for these compounds. Furthermore, the bacteriostasis achieved through physical or chemical principles in most of them, to a large extent, prevents the bacteria from obtaining drug resistance through mutation.
At present, the small molecule drugs are still adopted as the first choice of clinical antibacterial drugs, which, however, will bring some issues, as for example, they all act as the metabolic pathways shared by microorganisms, rather than drugs killing specific pathogenic bacteria, thus destroying the microbial ecological balance of the organism [21]. Among the novel treatment methods, antimicrobial therapy based on phage therapy and CRISPR-Cas technology has aroused the increasing interest of researchers. As early as 1921, Bruynoghe and Maisin [22][23] firstly applied phage preparations in treating skin infections caused by Staphylococci. spp. Since then, phages have been widely adopted in the treatment of otolaryngology, stomatology, ophthalmology, dermatology, and lung diseases. Despite the fact that after the 1940s, phage therapy had faded out due to the gradual popularization of antibiotics, it has returned as a part of the clinician’s weaponry with the increasingly serious problems of antibiotic resistance and the development of new antibiotics falling far behind in recent years, with multiple successful cases of superbugs treated by bacteriophages having been reported in clinical practice [24][25][26]. CRISPER-Cas, as a new gene editing technology, has a high targeting efficiency and simple primer design and multiple other advantages, which are utilized to specifically kill target pathogenic bacteria or knock out drug-resistant genes in the genome. They target and cut the precise sequences in the bacterial genome in a species-specific way to produce the narrowest antimicrobial spectrum possible, so as to achieve targeted sterilization or bacteriostasis. Both methods have developed to be one of the hot issues in the current research on pathogenic bacteria treatment.
2. Mechanism of Antibiotics Resistance
Plenty of antibiotic-resistant bacteria have continuously entered people’s vision since the discovery of the first antibiotic penicillin [12], triggering a long arms race between humans and bacteria. Despite the multiple natural and synthetic antibiotics that have been added to the battlefield, corresponding strategies will always be identified by bacteria to weaken the lethality of antibiotics. Moreover, additional functions also exist in a large amount of antibiotic resistance mechanisms in the metabolism process of bacteria. For instance, the efflux pump that transports specific antibiotics outside the cell membrane can also pump out toxins such as heavy metal ions to protect cells [27]. Facing the action of antibiotics, the related mechanisms have been continuously evolved by bacteria to resist antibiotics. Additionally, researchers have also discovered new resistance mechanisms to antibiotics in bacteria, involving entering the dormant state, secretion of target-protecting proteins, and regulation of metabolism and initiation of self-repair systems, which together constitute the bacterial defense system against antibiotics. Here, the mechanism of bacterial resistance to antibiotics is mainly summarized (Figure 1).
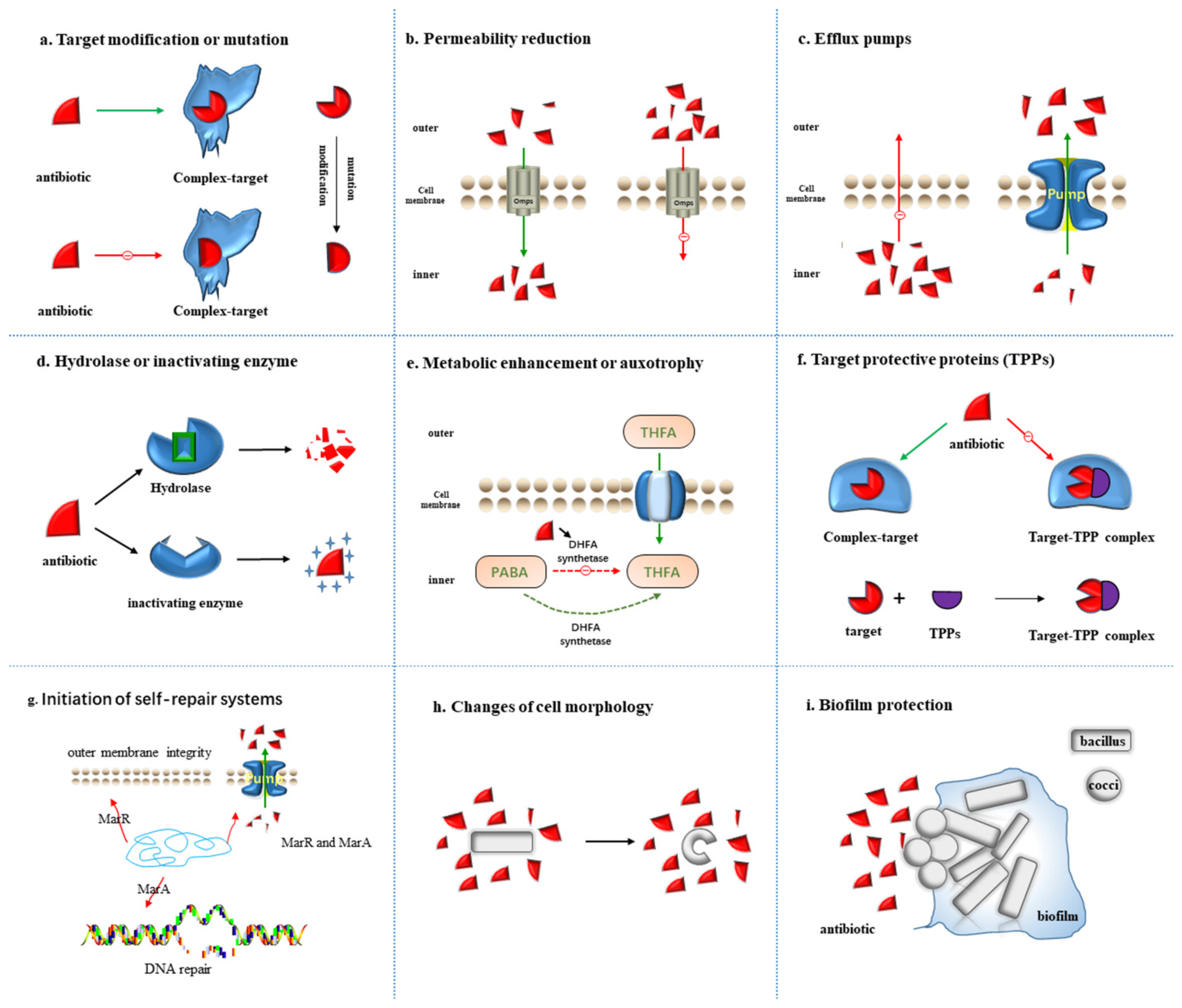
Figure 1. Nine resistance mechanisms of bacteria to antibiotics: (a) Target modification or mutation; (b) Permeability reduction; (c) Efflux pumps; (d) Hydrolase or inactivating enzyme; (e) Metabolic enhancement or auxotrophy; (f) Target protective protein; (g) Initiation of self-repair systems; (h) Changes of cell morphology; (i) Community cooperative resistance. The red triangle indicates antibiotics, and the resistance mechanisms of each figure are described in detail in the main text.
2.1. Target Modification or Mutation
Combination with the target site is required for antibiotics to exert an antibacterial effect, in which the mutation or modification of the target site will intervene with the normal combination, thus affecting the effect of antibiotics (Figure 1a). The frequency of spontaneous mutations in antibiotic resistance is about 10−8–10−9, which means that one in 108–109 bacteria will develop resistance through mutation [28]. The mutations occur randomly bound to the DNA replication process, and most are detrimental to the host bacteria, which will not be inherited at the cellular or population level. However, when exhibiting evolutionary advantage, the mutations may develop to be dominant through horizontal or vertical transmission. With the huge number of bacteria and the horrendous reproduction speed, the mutation of genes will correspondingly exhibit a high frequency, which allows bacteria to quickly acquire drug resistance through population evolution and horizontal gene transmission (HGT).
At present, it is a consensus that the resistance of Mycobacterium tuberculosis (MTB) to rifampicin (RFP) is mainly caused by the mutation of MTB rpoB gene [29]. The rpoB gene contains an open reading frame of 3534bp and encodes 1178 amino acids (AA), but many experimental studies have confirmed that its mutation mainly occurs in the 81-base region of position 507–533, which is called rifampicin resistance determining region (RRDR) [30][31]. However, some scholars believe that the mutation of RRDR external sequence plays a critical role in the resistance of MTB to RFP [32][33]. Penicillin-Binding Proteins (PBPs) [34] are located on the bacterial cytoplasmic membrane with roles in the synthesis of cell wall peptidoglycan, acting as the target of β-lactam antibiotics. When the mutation occurs, the affinity between β-lactam antibiotics and their target PBPs will disappear, resulting in the failure of the antibiotics to bind to the target, inducing bacterial resistance. A related example of target site modification is the structural alteration of PBPs in MRSA, where the resistance of S. aureus to methicillin results from the acquisition of an exogenous gene encoding PBP2a, called mecA, which is considered to have low affinity for most β-lactams as a PBPs enzyme. Thus, acquiring mecA renders most β-lactam antibiotics ineffective against MRSA. The similar target modification is also showed in bacteria resistance to vancomycin, macrolides, lincosamides, and streptavidin antibiotics [35].
2.2. Permeability Reduction
In Gram-negative bacteria (GNB), the cell wall is mainly constituted of proteins and lipopolysaccharides, in which the hydrophilic compounds are hard to pass through the lipid bilayer and must be facilitated by porin channels or outer membrane porins (Omps) [36][37]. Each type of bacteria produces specific porins (e.g., OmpF, OmpC, and OmpE), and the deletion or damage of one or more Omps is one of the sources for bacterial resistance [38]. For example, the loss of OprD porin on the outer membrane of the cell elicits the inefficiency or weakness against P. aeruginosa for many broad-spectrum antibacterial drugs, with which the antibacterial drugs cannot enter the cell, leading to the natural resistance to antibiotics (Figure 1b) [39].
After the exposure to antibiotics, the acquired drug resistance can be produced by changing the properties and quantity of porin to reduce the membrane permeability of bacteria. Normally, the channel proteins of bacterial outer membrane constitute non-specific transmembrane channels with OmpF and OmpC, allowing antibiotic and other drug molecules to enter the bacteria [40][41][42]. However, when bacteria are exposed to antibiotics more often, the mutations will be induced in the structural gene encoding OmpF protein, resulting in the reduction in, or loss of, OmpF channel protein, thus preventing the antibiotics such as β-lactams or quinolones to enter the bacteria normally. Gram-positive bacteria have no outer membrane to restrict the entry of drugs, and the outer membrane of mycobacteria is equipped with high lipid content, making the hydrophobic drugs (such as rifampicin and fluoroquinolone) easier to enter cells, while limiting the entry of hydrophilic drugs. Dong et al. [43] found that most strains with a higher resistance to β-lactam are accompanied by mutations of the OmpF-related gene of the membrane channel protein. The inactivation of the structural gene of the OmpF protein can decrease the membrane permeability of bacteria, intervening with the β-lactams, quinolones and other drugs to enter the bacteria, resulting in the acquired drug resistance.
2.3. Efflux Pumps
Depending on the antibiotic or toxin challenge, efflux may serve as the most rapid acting and most effective resistance mechanism in the bacterial repertoire of stress responses [44]. The bacterial efflux pump system [45] that has evolved in bacteria is a self-protection mechanism to prevent the accumulation of toxic compounds in cells, which can pump these harmful molecules out of the bacteria (Figure 1c). Bacterial efflux pumps (Eps) located in the plasma membrane of bacteria serve as the transporters to actively expel various substrates from the cytoplasm [46]. Among various families of transporters, several involve the prominent members of efflux transporters: the RND (resistance nodulation and cell division) that are especially crucial in bacteria; MFS (major facilitator superfamily); MATE (multidrug and toxic compound extrusion); SMR (small multidrug resistance); and ABC (ATP-binding cassette) superfamilies or families [47][48]. ABC efflux pumps (recognized as “primary active transporters”) eliminate substrates by consuming the energy generated by ATP hydrolysis, while “secondary active transporters” (MATE, MFS, RND and SMR) utilize proton motive force (PMF) as an energy source by pumping Na and hydrogen out of the membrane [49].
Currently, the efflux pumps identified in Gram-positive bacteria involve members of the MATE family and MFS family, where the MFS family is a characteristic efflux pump [50][51]. Efflux pumps identified in GNBare are widely distributed and may source from all the five families, with the most significant pumps in the clinic belonging to the RND family [44]. The RND efflux family members existing in many GNBand are involved in the efflux of antibiotics, heavy metals, toxins and many other substrates, some of which are specific, for example, Tet pumps tetracyclines or Mef pumps macrolides. Other RND pumps are mostly capable of delivering a wide range of drugs, such as the MexAB-OprM pump in P. aeruginosa, which confers intrinsic resistance to β-lactams, chloramphenicol, tetracycline, trimethoprim, sulfamethoxazole, and some fluorine Quinolones [52].
A lot of active efflux systems are nonspecific, which leads to multidrug resistance. For example, the active efflux system (AcorAB-TolC) of E. coli can elicit resistance to tetracycline, florfenicol, erythromycin, enrofloxacin and so on. Multiple efflux pump families have been identified in a strain of bacteria, with multiple members involved in each family. For example, the enterobacteriaceae is found to contain the RND efflux family, MFS efflux family and ABC efflux family. The RND efflux family involves AcrAB–TolC and OqxABa –TolC efflux systems, possessing the capability to efflux a variety of antibiotics [53][54]. For more detailed information on efflux pumps, such as the structure and function, it is recommended to refer to the review of DU’s Multidrug efflux pumps: structure, function and regulation [44].
2.4. Hydrolase or Inactivating Enzyme
The inactivating enzymes produced by bacteria, such as antibiotic hydrolases or inactivating enzymes, can hydrolyze or modify antibiotics entering the cell to render them inactive before reaching the target site (Figure 1d). There exist plenty of aminoglycoside-modifying enzymes in bacteria, such as N-acetyltransferase, O-phosphotransferase and O-adenosyltransferase, which, respectively, acetylate, phosphorylate or adenylate aminoglycoside antibiotics to transform the structure of antibiotics. Inactivating enzymes produced by bacteria mainly involve: β-lactamase, aminoglycoside inactivating enzymes, chloramphenicol acetyltransferase, etc. [55]. The β-lactamase can covalently bind to the carbonyl moiety of the antibiotic to disrupt its cyclic structure, inducing degradation in the β-lactam antibiotic before reaching the target. It can also rapidly and firmly bind to β-lactam antibiotics through non-hydrolysis, preventing the antibiotics from exerting drug resistance through binding to the target site. β-lactamases are secreted by many bacteria for up to eight different types, each capable of hydrolyzing specific β-lactam rings [56]. Carbapenem and extended-spectrum β-lactamases (ESBLs) are the two most primary β-lactamases. The main mechanism of Enterobacter resistance to carbapenem antibiotics is the production of enzymes that hydrolyze carbapenem. Those Enterobacteriaceae bacteria that can produce carbapenem enzymes are called carbapenem-producing Enterobacteriaceae (CPE). ESBLs can destroy most β-lactam antibiotics, such as penicillin and cephalosporins, but fail to destroy carbapenem antibiotics, and are commonly produced by E. coli, K. pneumoniae, P. aeruginosa, A. baumannii and other bacteria [57][58].
Rifampicin is adopted as the first choice for the treatment of tuberculosis and leprosy, of which the antibacterial activity depends on the inhibition of bacterial RNA polymerase. Researchers have identified a group of NAD-dependent enzymes in bacteria, which inactivate rifampicin [59] by transferring an ADP-ribosyl molecule to the hydroxyl group of the long aliphatic carbon chain of the rifampicin structure. Chloramphenicol acetyltransferases (CATs) promoted the acetyl group in acetyl-CoA to covalently link to two hydroxyl groups of chloramphenicol and prevent chloramphenicol from binding to ribosomes, thus exhibiting resistance to chloramphenicol [55]. Aminoglycosides play an antibacterial role by binding to 23SrRNA of bacterial 50S subunit to block protein synthesis. However, several resistance genes have been identified in S. aureus, Enterococcus faecium, M. tuberculosis, E. coli, Salmonella. spp. and other bacteria, such as lnu (A) to lnu (F) and linAN2, which inactivate lincomycin with the encoded nucleotide transferase [60].
2.5. Metabolic Alteration or Auxotrophy
Although metabolism has been demonstrated to actively contribute to antibiotic lethality, antibiotic resistance mutations are merely identified in metabolic genes, and metabolic dysregulation does not serve as a commonly cited mechanism of antibiotic resistance. In 2021, James’s team found for the first time that mutations in core genes in some metabolic pathways can induce antibiotic resistance, which are widely present in the genome of clinically pathogenic E. coli [61], including the core genes of metabolic pathways, such as the sucA gene (2-oxoglutarate dehydrogenase enzyme) involved in catalyzing the tricarboxylic acid cycle. The gene with this mutation reduces basal respiration by inhibiting the activity of the tricarboxylic acid cycle elicited by antibiotics, avoiding the occurrence of metabolic toxicity, inhibiting the killing effect of antibiotics, and eventually leading to antibiotic resistance [62].
The essential metabolic pathways required to synthesize amino acids, nucleotides, vitamins, fatty acids or metabolic coenzymes at the genetic level are found to be lacking in auxotrophs [63][64][65]. Microbial communities are composed of cells with varying metabolic capacity, regularly including auxotrophs lacking essential metabolic pathways. In contrast to prototrophs that can flexibly switch between metabolite synthesis and uptake, the growth of auxotrophs is constitutively dependent on the extracellular availability of these metabolites [66][67]. Sulfonamides possess a similar structure to that of p-aminobenzoic acid (PABA), which inhibits the activity of dihydrofolate synthase and prevents folate metabolism by competing with PABA to bind to the active site of dihydrofolate synthase in the process of bacterial folate metabolism. As folic acid is the precursor of nucleic acid synthesis, its deficiency will hinder nucleic acid synthesis and inhibit bacterial growth and reproduction [68]. However, bacteria can weaken the inhibitory effect of sulfonamide antibiotics on folic acid metabolism through metabolism enhancement, and can also obtain folic acid from extracellular in an auxotrophic way to maintain normal metabolism (Figure 1e). In 2022, Markus Ralser’s [69] team have revealed a metabolically imprinted mechanism that links the presence of auxotrophs to an enhancement in metabolic interactions and gains in antimicrobial drug tolerance. Moreover, the elevated efflux activities reduce the intracellular drug concentrations, allowing cells to grow in the presence of drug levels above minimal inhibitory concentrations. These results indicate that auxotrophy is beneficial to alleviate the sensitivity of bacteria to antibiotics, thus reducing the antibacterial effect of antibiotics.
2.6. Target Protective Proteins (TPPs)
Bacterial synthetic protein protects some antibiotic targets from a combination of antibiotics, eliminating their bacteriostatic effects (Figure 1f), and Daniel N. Wilson’s team [70] divided target protection into three types according to the mode of action. In Type I target protection, the binding of tetracycline ribosomal protection proteins (TRPPs) to ribosomes can reverse the distorted ribosomal structure, evoking changes in ribosome configuration, and directly interfering with the interaction of tetracycline D-ring and 16S rRNA base C1054. Tetracycline class drugs cannot bind to it and dissociate from the 30S subunit of the binding site, thereby protecting the ribosome, in which 13 TRPPs classes have been identified [61][62]. In Type II target protection, antibiotics are indirectly removed by changes in target conformation. Mediated by antibiotic-resistant ABC-F proteins, this group of proteins is the primary source of clinical resistance to antimicrobials of ribosome 50S subunits, including lincomycins, macrolides, azadones, phenols, pleuromutilins, and stroopogramins of groups A and B [71][72][73]. Type III target protection proteins induce changes in target conformation so that antibiotic targets can also work in the state of binding to antibiotics. In recent years, clinically isolated S.aureus and other staphylococcus resistance to fusidic acid has increased significantly, mainly due to the level acquisition of genes encoding the FusB-type protein. The resistance of FusB resistance proteins to fusidic acid is due to the fact that fusB proteins bind to elongation factor G (EF-G) and drive its dissociation from ribosomes (even in the presence of fusidic acid). Once the elongation factor leaves the ribosome, fusidic acid may be separated from EF-G due to its low affinity for free EF-G [74][75][76].
2.7. Initiation of Self-Repair Systems
The multiple antibiotic resistance operon of enteric bacteria manipulates the DNA repair and outer membrane integrity (Figure 1g), which contributes to enhancing the antibiotic resistance. The E. coli multiple antibiotic resistance (mar) locus was recognized as a determinant for cross-resistance to tetracyclines, quinolones and β-lactams [77]. Studies have shown that the active efflux mechanism controlled by the global operon is one of the primary reasons for the multiple antibiotic resistance of bacteria. Among them, the multiple antibiotic resistance protein family (Mar family), as a transcriptional regulatory protein, plays an important role in the production of drug resistance, the synthesis of toxic factors and other physiological processes. As the prototype of a multiple antibiotic resistance protein family, E. coli MarR protein has a negative regulatory function on MarRAB operon and inhibits the expression of downstream related drug resistance genes [78]. Transcription factors MarR and MarA confer multidrug resistance in enteric bacteria by modulating the efflux pump and porin expression [79][80][81]. In 2017, Sharma’s team demonstrated that MarA upregulates genes required for lipid trafficking and DNA repair, thus reducing DNA damage induced by antibiotic entry and quinolone [82]. The initiation of self-repair systems reduces the rate of antibiotics entering cells and the impact on cell structure and metabolism through gene regulation of the expression of related genes. This method cannot completely eliminate the bacteriostatic effect of antibiotics, but can make bacteria enhance their tolerance to antibiotics.
2.8. Changes of Cell Morphology
The mechanism of antibiotics is adapted through mechanical feedback between cell growth and morphology, altering uptake efficiency by modulating relative body area (Figure 1h). The increase in cell volume contributes to diluting the antibiotics entering the bacteria, while both bending and widening can reduce the surface volume ratio so that fewer antibiotics pass through its surface. In 2021, Aaron’s team [83] found that cells of the commonly used model organism C. crescentus could regain the growth rates they had prior to stimulation by antibiotics, accompanied with significant morphological transformations. Once the antibiotic was removed, the cells returned to their original shape after a few generations. That bacteria change shape to avoid being targeted by antibiotics was also previously demonstrated by another team [84], in which, however, the bacteria sloughed off their entire cell wall to avoid the drug, resulting in a shape distortion. In Aaron’s study, the cell walls remained intact, but stretched so violently that a “C” shape was formed. Bacteria are able to decrease the time it takes for antibiotics to exert biological effects in this physical way and increase the concentration of antibiotic tolerance. Using single-cell experiments and theoretical models, they proved that the change of cell morphology is a feedback strategy to enable it to adapt to the antibiotic environment and survive. The bacteria after “Metamorphosis” can overcome the pressure of antibiotics and recover to the state of rapid growth [83].
2.9. Biofilm Protection
Eight different drug resistance mechanisms of bacteria at the individual level have been summarized. However, in the actual environment, the vast majority of bacteria coexist in the form of communities, jointly resisting the effects of antibiotics in a collective form, with the biofilm serving as a critical form of protection (Figure 1h). Bacterial biofilm [85] is a special survival form established by bacteria adsorbed to inert objects such as medical materials or the surface of the body’s mucosa, in which the protein is surrounded by an autocrine polymer matrix. Dense biofilms are constituted to provide exposure protection for their members, forming physical barriers to limit the diffusion of antibiotics into the population and enhance the protection provided by antibiotic inactivation. In addition, due to the gradient of nutrients and oxygen, the decrease in the metabolic activity of the biofilm’s center enables the biofilm to induce a tolerant cell state, thus elevating the proportion of persistent cells in the population. Biofilms can also enhance the drug resistance by altering the expression of pre-existing ARG [86]. In contrast to single-species biofilms, the inter-species interactions among multi-species biofilms can further enhance the collective by altering the spatial structure of biofilms, promoting the expression of resistance mechanisms and allowing individually expressed antimicrobial defenses to protect entire communities. The resistance mechanism formed in the way of antibiotic resistance, collective tolerance or exposure protection to antibiotics is not specific and serves as the first line of defense for bacteria to develop resistance to antibiotics.
Bacterial communities can survive antibiotic exposure through interspecific interaction: (1) collective drug resistance, that is, the interaction within the community can enhance the capability of its members to resist antibiotics to continue to grow, thus elevating the MIC of the community; (2) Collective tolerance, i.e., interactions within the community can alter cellular states, such as retarding metabolism, so as to temporarily reduce the rate of cell death during antibiotic treatment without increasing the MIC; (3) Contact protection to protect the interaction of its sensitive members by reducing the effective concentration of antibiotics in the community [87][88]. In the mixed biofilm, P. aeruginosa can elicit the metabolic transformation of S. aureus, inhibit its growth and provide S. aureus with protection against vancomycin [89]. Correspondingly, S. aureus can enhance the tolerance of P. aeruginosa to tobramycin by promoting aggregation and altering the biofilm structure in the CF model system [90]; The interspecific signal transduction of indole secreted by E. coli activates the expression of indole dependent multidrug efflux pump in P. putida, while Pseudomonas itself cannot produce indole, which results in an elevation in the resistance level of P. putida [91]. Similarly, S. maltophilia is a Gram-negative bacterium, generally appearing accompanied by P. aeruginosa during bacterial lung infection. It can diffuse the secretion of signal factors, transform the biofilm structure of P. aeruginosa, and stimulate the synthesis of proteins so as to provide resistance to cationic antimicrobial peptides, such as polymyxin [92].
References
- Roope, L.S.J.; Smith, R.D.; Pouwels, K.B.; Buchanan, J.; Abel, L.; Eibich, P.; Butler, C.C.; Tan, P.S.; Walker, A.S.; Robotham, J.V.; et al. The challenge of antimicrobial resistance: What economics can contribute. Science 2019, 364, eaau4679.
- Theuretzbacher, U.; Gottwalt, S.; Beyer, P.; Butler, M.; Czaplewski, L.; Lienhardt, C.; Moja, L.; Paul, M.; Paulin, S.; Rex, J.H.; et al. Analysis of the clinical antibacterial and antituberculosis pipeline. Lancet Infect. Dis. 2019, 19, e40–e50.
- Zhang, Q.Q.; Ying, G.G.; Pan, C.G.; Liu, Y.S.; Zhao, J.L. Comprehensive evaluation of antibiotics emission and fate in the river basins of China: Source analysis, multimedia modeling, and linkage to bacterial resistance. Env. Sci. Technol. 2015, 49, 6772–6782.
- Antimicrobial Resistance, C. Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 2022, 399, 629–655.
- Diseases, G.B.D.; Injuries, C. Global burden of 369 diseases and injuries in 204 countries and territories, 1990-2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet 2020, 396, 1204–1222.
- Fleming, A. On the antibacterial action of cultures of a penicillium, with special reference to their use in the isolation of B. influenzae. 1929. Bull. World Health Organ. 2001, 79, 780–790.
- Chain, E.; Florey, H.W.; Gardner, A.D.; Heatley, N.G.; Jennings, M.A.; Orr-Ewing, J.; Sanders, A.G. Penicillin as a chemotherapeutic agent. Lancet 1940, 236, 226–228.
- Lee, A.S.; de Lencastre, H.; Garau, J.; Kluytmans, J.; Malhotra-Kumar, S.; Peschel, A.; Harbarth, S. Methicillin-resistant Staphylococcus aureus. Nat. Reviews. Dis. Primers 2018, 4, 18033.
- Yong, D.; Toleman, M.A.; Giske, C.G.; Cho, H.S.; Sundman, K.; Lee, K.; Walsh, T.R. Characterization of a new metallo-beta-lactamase gene, bla(NDM-1), and a novel erythromycin esterase gene carried on a unique genetic structure in Klebsiella pneumoniae sequence type 14 from India. Antimicrob. Agents Chemother. 2009, 53, 5046–5054.
- Liu, Y.Y.; Wang, Y.; Walsh, T.R.; Yi, L.X.; Zhang, R.; Spencer, J.; Doi, Y.; Tian, G.; Dong, B.; Huang, X.; et al. Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: A microbiological and molecular biological study. Lancet. Infect. Dis. 2016, 16, 161–168.
- Who Publishes List Of Bacteria for Which New Antibiotics Are Urgently Needed. Saudi Med. J. 2017, 38, 444–445.
- Mohr, K.I. History of Antibiotics Research. Curr. Top Microbiol. Immunol. 2016, 398, 237–272.
- Lewis, K. The Science of Antibiotic Discovery. Cell 2020, 181, 29–45.
- D’Costa, V.M.; King, C.E.; Kalan, L.; Morar, M.; Sung, W.W.; Schwarz, C.; Froese, D.; Zazula, G.; Calmels, F.; Debruyne, R.; et al. Antibiotic resistance is ancient. Nature 2011, 477, 457–461.
- Lebreton, F.; Manson, A.L.; Saavedra, J.T.; Straub, T.J.; Earl, A.M.; Gilmore, M.S. Tracing the Enterococci from Paleozoic Origins to the Hospital. Cell 2017, 169, 849–861.e813.
- Shukla, R.; Medeiros-Silva, J.; Parmar, A.; Vermeulen, B.J.A.; Das, S.; Paioni, A.L.; Jekhmane, S.; Lorent, J.; Bonvin, A.; Baldus, M.; et al. Mode of action of teixobactins in cellular membranes. Nat. Commun. 2020, 11, 2848.
- Luther, A.; Urfer, M.; Zahn, M.; Muller, M.; Wang, S.Y.; Mondal, M.; Vitale, A.; Hartmann, J.B.; Sharpe, T.; Monte, F.L.; et al. Chimeric peptidomimetic antibiotics against Gram-negative bacteria. Nature 2019, 576, 452–458.
- Smith, P.A.; Koehler, M.F.T.; Girgis, H.S.; Yan, D.; Chen, Y.; Chen, Y.; Crawford, J.J.; Durk, M.R.; Higuchi, R.I.; Kang, J.; et al. Optimized arylomycins are a new class of Gram-negative antibiotics. Nature 2018, 561, 189–194.
- Culp, E.J.; Waglechner, N.; Wang, W.; Fiebig-Comyn, A.A.; Hsu, Y.P.; Koteva, K.; Sychantha, D.; Coombes, B.K.; Van Nieuwenhze, M.S.; Brun, Y.V.; et al. Evolution-guided discovery of antibiotics that inhibit peptidoglycan remodelling. Nature 2020, 578, 582–587.
- Mitcheltree, M.J.; Pisipati, A.; Syroegin, E.A.; Silvestre, K.J.; Klepacki, D.; Mason, J.D.; Terwilliger, D.W.; Testolin, G.; Pote, A.R.; Wu, K.J.Y.; et al. A synthetic antibiotic class overcoming bacterial multidrug resistance. Nature 2021, 599, 507–512.
- Dethlefsen, L.; Huse, S.; Sogin, M.L.; Relman, D.A. The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol. 2008, 6, e280.
- Trudil, D. Phage lytic enzymes: A history. Virol. Sin. 2015, 30, 26–32.
- Duzgunes, N.; Sessevmez, M.; Yildirim, M. Bacteriophage Therapy of Bacterial Infections: The Rediscovered Frontier. Pharmaceuticals 2021, 14, 34.
- Schooley, R.T.; Biswas, B.; Gill, J.J.; Hernandez-Morales, A.; Lancaster, J.; Lessor, L.; Barr, J.J.; Reed, S.L.; Rohwer, F.; Benler, S.; et al. Development and Use of Personalized Bacteriophage-Based Therapeutic Cocktails To Treat a Patient with a Disseminated Resistant Acinetobacter baumannii Infection. Antimicrob. Agents Chemother. 2017, 61, e00954-17.
- Nir-Paz, R.; Gelman, D.; Khouri, A.; Sisson, B.M.; Fackler, J.; Alkalay-Oren, S.; Khalifa, L.; Rimon, A.; Yerushalmy, O.; Bader, R.; et al. Successful Treatment of Antibiotic-resistant, Poly-microbial Bone Infection With Bacteriophages and Antibiotics Combination. Clin. Infect Dis. 2019, 69, 2015–2018.
- Eskenazi, A.; Lood, C.; Wubbolts, J.; Hites, M.; Balarjishvili, N.; Leshkasheli, L.; Askilashvili, L.; Kvachadze, L.; van Noort, V.; Wagemans, J.; et al. Combination of pre-adapted bacteriophage therapy and antibiotics for treatment of fracture-related infection due to pandrug-resistant Klebsiella pneumoniae. Nat. Commun. 2022, 13, 302.
- Du, D.; Wang-Kan, X.; Neuberger, A.; van Veen, H.W.; Pos, K.M.; Piddock, L.J.V.; Luisi, B.F. Multidrug efflux pumps: Structure, function and regulation. Nat. Rev. Microbiol. 2018, 16, 523–539.
- Davies, J. Origins and evolution of antibiotic resistance. Microbiologia 1996, 12, 9–16.
- Goldstein, B.P. Resistance to rifampicin: A review. J. Antibiot. 2014, 67, 625–630.
- Ahmad, S.; Mokaddas, E.; Fares, E. Characterization of rpoB mutations in rifampin-resistant clinical Mycobacterium tuberculosis isolates from Kuwait and Dubai. Diagn. Microbiol. Infect. Dis. 2002, 44, 245–252.
- Chen, L.; Gan, X.; Li, N.; Wang, J.; Li, K.; Zhang, H. rpoB gene mutation profile in rifampicin-resistant Mycobacterium tuberculosis clinical isolates from Guizhou, one of the highest incidence rate regions in China. J. Antimicrob. Chemother. 2010, 65, 1299–1301.
- Tan, Y.; Hu, Z.; Zhao, Y.; Cai, X.; Luo, C.; Zou, C.; Liu, X. The beginning of the rpoB gene in addition to the rifampin resistance determination region might be needed for identifying rifampin/rifabutin cross-resistance in multidrug-resistant Mycobacterium tuberculosis isolates from Southern China. J. Clin. Microbiol. 2012, 50, 81–85.
- Siu, G.K.; Zhang, Y.; Lau, T.C.; Lau, R.W.; Ho, P.L.; Yew, W.W.; Tsui, S.K.; Cheng, V.C.; Yuen, K.Y.; Yam, W.C. Mutations outside the rifampicin resistance-determining region associated with rifampicin resistance in Mycobacterium tuberculosis. J. Antimicrob. Chemother. 2011, 66, 730–733.
- Miyachiro, M.M.; Contreras-Martel, C.; Dessen, A. Penicillin-Binding Proteins (PBPs) and Bacterial Cell Wall Elongation Complexes. Subcell. Biochem. 2019, 93, 273–289.
- Miragaia, M. Factors Contributing to the Evolution of mecA-Mediated β-lactam Resistance in Staphylococci: Update and New Insights From Whole Genome Sequencing (WGS). Front. Microbiol. 2018, 9, 2723.
- Nikaido, H. Molecular basis of bacterial outer membrane permeability revisited. Microbiol. Mol. Biol. Rev. 2003, 67, 593–656.
- Chevalier, S.; Bouffartigues, E.; Bodilis, J.; Maillot, O.; Lesouhaitier, O.; Feuilloley, M.G.J.; Orange, N.; Dufour, A.; Cornelis, P. Structure, function and regulation of Pseudomonas aeruginosa porins. FEMS Microbiol. Rev. 2017, 41, 698–722.
- McPhee, J.B.; Tamber, S.; Brazas, M.D.; Lewenza, S.; Hancock, R.E.W. Antibiotic Resistance Due to Reduced Uptake. In Antimicrobial Drug Resistance: Mechanisms of Drug Resistance; Mayers, D.L., Ed.; Humana Press: Totowa, NJ, USA, 2009; pp. 97–110.
- Nadeem, S.F.; Gohar, U.F.; Tahir, S.F.; Mukhtar, H.; Pornpukdeewattana, S.; Nukthamna, P.; Moula Ali, A.M.; Bavisetty, S.C.B.; Massa, S. Antimicrobial resistance: More than 70 years of war between humans and bacteria. Crit. Rev. Microbiol. 2020, 46, 578–599.
- Moya-Torres, A.; Mulvey, M.R.; Kumar, A.; Oresnik, I.J.; Brassinga, A.K.C. The lack of OmpF, but not OmpC, contributes to increased antibiotic resistance in Serratia marcescens. Microbiol. (Read. Engl.) 2014, 160, 1882–1892.
- Ziervogel, B.K.; Roux, B. The Binding of Antibiotics in OmpF Porin. Structure 2013, 21, 76–87.
- Bafna, J.A.; Sans-Serramitjana, E.; Acosta-Gutiérrez, S.; Bodrenko, I.V.; Hörömpöli, D.; Berscheid, A.; Brötz-Oesterhelt, H.; Winterhalter, M.; Ceccarelli, M. Kanamycin Uptake into Escherichia coli Is Facilitated by OmpF and OmpC Porin Channels Located in the Outer Membrane. ACS Infect. Dis. 2020, 6, 1855–1865.
- Tang, X.; Chang, S.; Qiao, W.; Luo, Q.; Chen, Y.; Jia, Z.; Coleman, J.; Zhang, K.; Wang, T.; Zhang, Z.; et al. Structural insights into outer membrane asymmetry maintenance in Gram-negative bacteria by MlaFEDB. Nat. Struct. Mol. Biol. 2021, 28, 81–91.
- Rahman, T.; Yarnall, B.; Doyle, D.A. Efflux drug transporters at the forefront of antimicrobial resistance. Eur. Biophys. J. EBJ 2017, 46, 647–653.
- Amaral, L.; Martins, A.; Spengler, G.; Molnar, J. Efflux pumps of Gram-negative bacteria: What they do, how they do it, with what and how to deal with them. Front. Pharm. 2014, 4, 168.
- Blanco, P.; Hernando-Amado, S.; Reales-Calderon, J.A.; Corona, F.; Lira, F.; Alcalde-Rico, M.; Bernardini, A.; Sanchez, M.B.; Martinez, J.L. Bacterial Multidrug Efflux Pumps: Much More Than Antibiotic Resistance Determinants. Microorganisms 2016, 4, 14.
- Hernando-Amado, S.; Blanco, P.; Alcalde-Rico, M.; Corona, F.; Reales-Calderón, J.A.; Sánchez, M.B.; Martínez, J.L. Multidrug efflux pumps as main players in intrinsic and acquired resistance to antimicrobials. Drug Resist. Updates Rev. Comment. Antimicrob. Anticancer. Chemother. 2016, 28, 13–27.
- Li, X.Z.; Plesiat, P.; Nikaido, H. The challenge of efflux-mediated antibiotic resistance in Gram-negative bacteria. Clin. Microbiol. Rev. 2015, 28, 337–418.
- Kourtesi, C.; Ball, A.R.; Huang, Y.Y.; Jachak, S.M.; Vera, D.M.; Khondkar, P.; Gibbons, S.; Hamblin, M.R.; Tegos, G.P. Microbial efflux systems and inhibitors: Approaches to drug discovery and the challenge of clinical implementation. Open Microbiol. J. 2013, 7, 34–52.
- Costa, S.S.; Viveiros, M.; Amaral, L.; Couto, I. Multidrug Efflux Pumps in Staphylococcus aureus: An Update. Open Microbiol. J. 2013, 7, 59–71.
- Reygaert, W.C. An overview of the antimicrobial resistance mechanisms of bacteria. AIMS Microbiol. 2018, 4, 482–501.
- Swick, M.C.; Morgan-Linnell, S.K.; Carlson, K.M.; Zechiedrich, L. Expression of multidrug efflux pump genes acrAB-tolC, mdfA, and norE in Escherichia coli clinical isolates as a function of fluoroquinolone and multidrug resistance. Antimicrob. Agents Chemother. 2011, 55, 921–924.
- Hansen, L.H.; Jensen, L.B.; Sorensen, H.I.; Sorensen, S.J. Substrate specificity of the OqxAB multidrug resistance pump in Escherichia coli and selected enteric bacteria. J. Antimicrob. Chemother. 2007, 60, 145–147.
- Peterson, E.; Kaur, P. Antibiotic Resistance Mechanisms in Bacteria: Relationships Between Resistance Determinants of Antibiotic Producers, Environmental Bacteria, and Clinical Pathogens. Front. Microbiol. 2018, 9, 2928.
- Tooke, C.L.; Hinchliffe, P.; Bragginton, E.C.; Colenso, C.K.; Hirvonen, V.H.A.; Takebayashi, Y.; Spencer, J. β-Lactamases and β-Lactamase Inhibitors in the 21st Century. J. Mol. Biol. 2019, 431, 3472–3500.
- Ghafourian, S.; Sadeghifard, N.; Soheili, S.; Sekawi, Z. Extended Spectrum Beta-lactamases: Definition, Classification and Epidemiology. Curr. Issues Mol. Biol. 2015, 17, 11–21.
- Sawa, T.; Kooguchi, K.; Moriyama, K. Molecular diversity of extended-spectrum β-lactamases and carbapenemases, and antimicrobial resistance. J. Intensive Care 2020, 8, 13.
- Sköld, O. Aminoglycosides. In Antibiotics and Antibiotic Resistance; John Wiley & Sons: Hoboken, NJ, USA, 2011; Volume 6, pp. 103–113.
- Schwarz, S.; Shen, J.; Kadlec, K.; Wang, Y.; Brenner Michael, G.; Fessler, A.T.; Vester, B. Lincosamides, Streptogramins, Phenicols, and Pleuromutilins: Mode of Action and Mechanisms of Resistance. Cold Spring Harb. Perspect Med. 2016, 6, a027037.
- Lopatkin, A.J.; Bening, S.C.; Manson, A.L.; Stokes, J.M.; Kohanski, M.A.; Badran, A.H.; Earl, A.M.; Cheney, N.J.; Yang, J.H.; Collins, J.J. Clinically relevant mutations in core metabolic genes confer antibiotic resistance. Science 2021, 371, eaba0862.
- Zampieri, M. The genetic underground of antibiotic resistance. Science 2021, 371, 783–784.
- Mee, M.T.; Collins, J.J.; Church, G.M.; Wang, H.H. Syntrophic exchange in synthetic microbial communities. Proc. Natl. Acad. Sci. USA 2014, 111, E2149–E2156.
- Croft, M.T.; Lawrence, A.D.; Raux-Deery, E.; Warren, M.J.; Smith, A.G. Algae acquire vitamin B12 through a symbiotic relationship with bacteria. Nature 2005, 438, 90–93.
- Sieuwerts, S.; Molenaar, D.; van Hijum, S.A.; Beerthuyzen, M.; Stevens, M.J.; Janssen, P.W.; Ingham, C.J.; de Bok, F.A.; de Vos, W.M.; van Hylckama Vlieg, J.E. Mixed-culture transcriptome analysis reveals the molecular basis of mixed-culture growth in Streptococcus thermophilus and Lactobacillus bulgaricus. Appl. Env. Microbiol. 2010, 76, 7775–7784.
- Zelezniak, A.; Andrejev, S.; Ponomarova, O.; Mende, D.R.; Bork, P.; Patil, K.R. Metabolic dependencies drive species co-occurrence in diverse microbial communities. Proc. Natl. Acad. Sci. USA 2015, 112, 6449–6454.
- Seif, Y.; Choudhary, K.S.; Hefner, Y.; Anand, A.; Yang, L.; Palsson, B.O. Metabolic and genetic basis for auxotrophies in Gram-negative species. Proc. Natl. Acad. Sci. USA 2020, 117, 6264–6273.
- Sun, H.; Yao, Z.; Wang, D.; Wu, X.; Lin, Z.; Liu, Y. A deep insight into the toxic mechanism for sulfonamides based on bacterial cell-cell communication. Environ. Int. 2019, 129, 185–193.
- Yu, J.S.L.; Correia-Melo, C.; Zorrilla, F.; Herrera-Dominguez, L.; Wu, M.Y.; Hartl, J.; Campbell, K.; Blasche, S.; Kreidl, M.; Egger, A.S.; et al. Microbial communities form rich extracellular metabolomes that foster metabolic interactions and promote drug tolerance. Nat. Microbiol. 2022, 7, 542–555.
- Wilson, D.N.; Hauryliuk, V.; Atkinson, G.C.; O’Neill, A.J. Target protection as a key antibiotic resistance mechanism. Nat. Rev. Microbiol. 2020, 18, 637–648.
- Sharkey, L.K.; Edwards, T.A.; O’Neill, A.J. ABC-F Proteins Mediate Antibiotic Resistance through Ribosomal Protection. mBio 2016, 7, e01975.
- Sharkey, L.K.R.; O’Neill, A.J. Antibiotic Resistance ABC-F Proteins: Bringing Target Protection into the Limelight. ACS Infect. Dis. 2018, 4, 239–246.
- Su, W.; Kumar, V.; Ding, Y.; Ero, R.; Serra, A.; Lee, B.S.T.; Wong, A.S.W.; Shi, J.; Sze, S.K.; Yang, L.; et al. Ribosome protection by antibiotic resistance ATP-binding cassette protein. Proc. Natl. Acad. Sci. USA 2018, 115, 5157–5162.
- Seo, H.S.; Abedin, S.; Kamp, D.; Wilson, D.N.; Nierhaus, K.H.; Cooperman, B.S. EF-G-dependent GTPase on the ribosome. conformational change and fusidic acid inhibition. Biochemistry 2006, 45, 2504–2514.
- Castanheira, M.; Watters, A.A.; Mendes, R.E.; Farrell, D.J.; Jones, R.N. Occurrence and molecular characterization of fusidic acid resistance mechanisms among Staphylococcus spp. from European countries (2008). J. Antimicrob. Chemother. 2010, 65, 1353–1358.
- McLaws, F.; Chopra, I.; O’Neill, A.J. High prevalence of resistance to fusidic acid in clinical isolates of Staphylococcus epidermidis. J. Antimicrob. Chemother. 2008, 61, 1040–1043.
- George, A.M.; Levy, S.B. Amplifiable resistance to tetracycline, chloramphenicol, and other antibiotics in Escherichia coli: Involvement of a non-plasmid-determined efflux of tetracycline. J. Bacteriol. 1983, 155, 531–540.
- Hao, Z.; Lou, H.; Zhu, R.; Zhu, J.; Zhang, D.; Zhao, B.S.; Zeng, S.; Chen, X.; Chan, J.; He, C.; et al. The multiple antibiotic resistance regulator MarR is a copper sensor in Escherichia coli. Nat. Chem. Biol. 2014, 10, 21–28.
- Ariza, R.R.; Cohen, S.P.; Bachhawat, N.; Levy, S.B.; Demple, B. Repressor mutations in the marRAB operon that activate oxidative stress genes and multiple antibiotic resistance in Escherichia coli. J. Bacteriol. 1994, 176, 143–148.
- McMurry, L.M.; George, A.M.; Levy, S.B. Active efflux of chloramphenicol in susceptible Escherichia coli strains and in multiple-antibiotic-resistant (Mar) mutants. Antimicrob. Agents Chemother. 1994, 38, 542–546.
- Zhang, A.; Rosner, J.L.; Martin, R.G. Transcriptional activation by MarA, SoxS and Rob of two tolC promoters using one binding site: A complex promoter configuration for tolC in Escherichia coli. Mol. Microbiol. 2008, 69, 1450–1455.
- Sharma, P.; Haycocks, J.R.J.; Middlemiss, A.D.; Kettles, R.A.; Sellars, L.E.; Ricci, V.; Piddock, L.J.V.; Grainger, D.C. The multiple antibiotic resistance operon of enteric bacteria controls DNA repair and outer membrane integrity. Nat. Commun. 2017, 8, 1444.
- Banerjee, S.; Lo, K.; Ojkic, N.; Stephens, R.; Scherer, N.F.; Dinner, A.R. Mechanical feedback promotes bacterial adaptation to antibiotics. Nat. Phys. 2021, 17, 403–409.
- Mickiewicz, K.M.; Kawai, Y.; Drage, L.; Gomes, M.C.; Davison, F.; Pickard, R.; Hall, J.; Mostowy, S.; Aldridge, P.D.; Errington, J. Possible role of L-form switching in recurrent urinary tract infection. Nat. Commun. 2019, 10, 4379.
- Tolker-Nielsen, T. Biofilm Development. Microbiol. Spectr. 2015, 3, MB-0001-2014.
- Høiby, N.; Bjarnsholt, T.; Givskov, M.; Molin, S.; Ciofu, O. Antibiotic resistance of bacterial biofilms. Int. J. Antimicrob. Agents 2010, 35, 322–332.
- Bottery, M.J.; Pitchford, J.W.; Friman, V.P. Ecology and evolution of antimicrobial resistance in bacterial communities. ISME J. 2021, 15, 939–948.
- Vega, N.M.; Gore, J. Collective antibiotic resistance: Mechanisms and implications. Curr. Opin. Microbiol. 2014, 21, 28–34.
- Orazi, G.; O’Toole, G.A. Pseudomonas aeruginosa Alters Staphylococcus aureus Sensitivity to Vancomycin in a Biofilm Model of Cystic Fibrosis Infection. mBio 2017, 8, e00873-17.
- Beaudoin, T.; Yau, Y.C.W.; Stapleton, P.J.; Gong, Y.; Wang, P.W.; Guttman, D.S.; Waters, V. Staphylococcus aureus interaction with Pseudomonas aeruginosa biofilm enhances tobramycin resistance. NPJ Biofilms Microbiomes 2017, 3, 25.
- Molina-Santiago, C.; Daddaoua, A.; Fillet, S.; Duque, E.; Ramos, J.L. Interspecies signalling: Pseudomonas putida efflux pump TtgGHI is activated by indole to increase antibiotic resistance. Environ. Microbiol. 2014, 16, 1267–1281.
- Ryan, R.P.; Fouhy, Y.; Garcia, B.F.; Watt, S.A.; Niehaus, K.; Yang, L.; Tolker-Nielsen, T.; Dow, J.M. Interspecies signalling via the Stenotrophomonas maltophilia diffusible signal factor influences biofilm formation and polymyxin tolerance in Pseudomonas aeruginosa. Mol. Microbiol. 2008, 68, 75–86.
- Ling, L.L.; Schneider, T.; Peoples, A.J.; Spoering, A.L.; Engels, I.; Conlon, B.P.; Mueller, A.; Schäberle, T.F.; Hughes, D.E.; Epstein, S.; et al. A new antibiotic kills pathogens without detectable resistance. Nature 2015, 517, 455–459.
More
Information
Subjects:
Pharmacology & Pharmacy
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.4K
Revisions:
2 times
(View History)
Update Date:
28 Sep 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No