Bacterial drug resistance is rapidly developing as one of the greatest threats to human health. Bacteria will adopt corresponding strategies to crack the inhibitory effect of antibiotics according to the antibacterial mechanism of antibiotics, involving the mutation of drug target, secreting hydrolase, and discharging antibiotics out of cells through an efflux pump, etc. Bacteria are found to constantly evolve new resistance mechanisms to antibiotics, including target protective protein, changes in cell morphology, and so on, endowing them with multiple defense systems against antibiotics, leading to the emergence of multi-drug resistant (MDR) bacteria and the unavailability of drugs in clinics.
- bacterial drug resistance
- antibacterial compounds
1. Introduction
2. Mechanism of Antibiotics Resistance
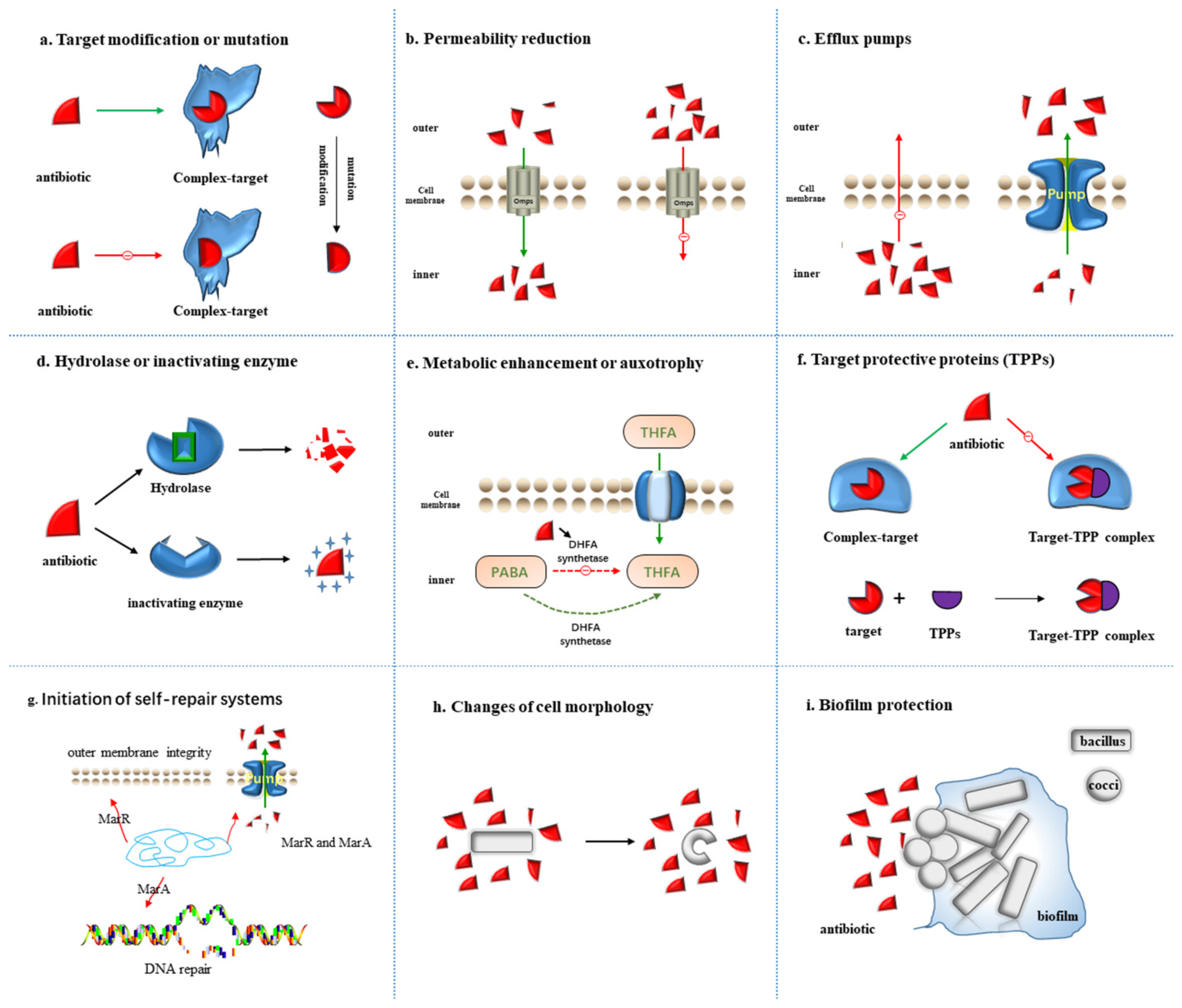
2.1. Target Modification or Mutation
2.2. Permeability Reduction
2.3. Efflux Pumps
2.4. Hydrolase or Inactivating Enzyme
2.5. Metabolic Alteration or Auxotrophy
2.6. Target Protective Proteins (TPPs)
2.7. Initiation of Self-Repair Systems
2.8. Changes of Cell Morphology
2.9. Biofilm Protection
This entry is adapted from the peer-reviewed paper 10.3390/antibiotics11091215
References
- Roope, L.S.J.; Smith, R.D.; Pouwels, K.B.; Buchanan, J.; Abel, L.; Eibich, P.; Butler, C.C.; Tan, P.S.; Walker, A.S.; Robotham, J.V.; et al. The challenge of antimicrobial resistance: What economics can contribute. Science 2019, 364, eaau4679.
- Theuretzbacher, U.; Gottwalt, S.; Beyer, P.; Butler, M.; Czaplewski, L.; Lienhardt, C.; Moja, L.; Paul, M.; Paulin, S.; Rex, J.H.; et al. Analysis of the clinical antibacterial and antituberculosis pipeline. Lancet Infect. Dis. 2019, 19, e40–e50.
- Zhang, Q.Q.; Ying, G.G.; Pan, C.G.; Liu, Y.S.; Zhao, J.L. Comprehensive evaluation of antibiotics emission and fate in the river basins of China: Source analysis, multimedia modeling, and linkage to bacterial resistance. Env. Sci. Technol. 2015, 49, 6772–6782.
- Antimicrobial Resistance, C. Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 2022, 399, 629–655.
- Diseases, G.B.D.; Injuries, C. Global burden of 369 diseases and injuries in 204 countries and territories, 1990-2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet 2020, 396, 1204–1222.
- Fleming, A. On the antibacterial action of cultures of a penicillium, with special reference to their use in the isolation of B. influenzae. 1929. Bull. World Health Organ. 2001, 79, 780–790.
- Chain, E.; Florey, H.W.; Gardner, A.D.; Heatley, N.G.; Jennings, M.A.; Orr-Ewing, J.; Sanders, A.G. Penicillin as a chemotherapeutic agent. Lancet 1940, 236, 226–228.
- Lee, A.S.; de Lencastre, H.; Garau, J.; Kluytmans, J.; Malhotra-Kumar, S.; Peschel, A.; Harbarth, S. Methicillin-resistant Staphylococcus aureus. Nat. Reviews. Dis. Primers 2018, 4, 18033.
- Yong, D.; Toleman, M.A.; Giske, C.G.; Cho, H.S.; Sundman, K.; Lee, K.; Walsh, T.R. Characterization of a new metallo-beta-lactamase gene, bla(NDM-1), and a novel erythromycin esterase gene carried on a unique genetic structure in Klebsiella pneumoniae sequence type 14 from India. Antimicrob. Agents Chemother. 2009, 53, 5046–5054.
- Liu, Y.Y.; Wang, Y.; Walsh, T.R.; Yi, L.X.; Zhang, R.; Spencer, J.; Doi, Y.; Tian, G.; Dong, B.; Huang, X.; et al. Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: A microbiological and molecular biological study. Lancet. Infect. Dis. 2016, 16, 161–168.
- Who Publishes List Of Bacteria for Which New Antibiotics Are Urgently Needed. Saudi Med. J. 2017, 38, 444–445.
- Mohr, K.I. History of Antibiotics Research. Curr. Top Microbiol. Immunol. 2016, 398, 237–272.
- Lewis, K. The Science of Antibiotic Discovery. Cell 2020, 181, 29–45.
- D’Costa, V.M.; King, C.E.; Kalan, L.; Morar, M.; Sung, W.W.; Schwarz, C.; Froese, D.; Zazula, G.; Calmels, F.; Debruyne, R.; et al. Antibiotic resistance is ancient. Nature 2011, 477, 457–461.
- Lebreton, F.; Manson, A.L.; Saavedra, J.T.; Straub, T.J.; Earl, A.M.; Gilmore, M.S. Tracing the Enterococci from Paleozoic Origins to the Hospital. Cell 2017, 169, 849–861.e813.
- Shukla, R.; Medeiros-Silva, J.; Parmar, A.; Vermeulen, B.J.A.; Das, S.; Paioni, A.L.; Jekhmane, S.; Lorent, J.; Bonvin, A.; Baldus, M.; et al. Mode of action of teixobactins in cellular membranes. Nat. Commun. 2020, 11, 2848.
- Luther, A.; Urfer, M.; Zahn, M.; Muller, M.; Wang, S.Y.; Mondal, M.; Vitale, A.; Hartmann, J.B.; Sharpe, T.; Monte, F.L.; et al. Chimeric peptidomimetic antibiotics against Gram-negative bacteria. Nature 2019, 576, 452–458.
- Smith, P.A.; Koehler, M.F.T.; Girgis, H.S.; Yan, D.; Chen, Y.; Chen, Y.; Crawford, J.J.; Durk, M.R.; Higuchi, R.I.; Kang, J.; et al. Optimized arylomycins are a new class of Gram-negative antibiotics. Nature 2018, 561, 189–194.
- Culp, E.J.; Waglechner, N.; Wang, W.; Fiebig-Comyn, A.A.; Hsu, Y.P.; Koteva, K.; Sychantha, D.; Coombes, B.K.; Van Nieuwenhze, M.S.; Brun, Y.V.; et al. Evolution-guided discovery of antibiotics that inhibit peptidoglycan remodelling. Nature 2020, 578, 582–587.
- Mitcheltree, M.J.; Pisipati, A.; Syroegin, E.A.; Silvestre, K.J.; Klepacki, D.; Mason, J.D.; Terwilliger, D.W.; Testolin, G.; Pote, A.R.; Wu, K.J.Y.; et al. A synthetic antibiotic class overcoming bacterial multidrug resistance. Nature 2021, 599, 507–512.
- Dethlefsen, L.; Huse, S.; Sogin, M.L.; Relman, D.A. The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol. 2008, 6, e280.
- Trudil, D. Phage lytic enzymes: A history. Virol. Sin. 2015, 30, 26–32.
- Duzgunes, N.; Sessevmez, M.; Yildirim, M. Bacteriophage Therapy of Bacterial Infections: The Rediscovered Frontier. Pharmaceuticals 2021, 14, 34.
- Schooley, R.T.; Biswas, B.; Gill, J.J.; Hernandez-Morales, A.; Lancaster, J.; Lessor, L.; Barr, J.J.; Reed, S.L.; Rohwer, F.; Benler, S.; et al. Development and Use of Personalized Bacteriophage-Based Therapeutic Cocktails To Treat a Patient with a Disseminated Resistant Acinetobacter baumannii Infection. Antimicrob. Agents Chemother. 2017, 61, e00954-17.
- Nir-Paz, R.; Gelman, D.; Khouri, A.; Sisson, B.M.; Fackler, J.; Alkalay-Oren, S.; Khalifa, L.; Rimon, A.; Yerushalmy, O.; Bader, R.; et al. Successful Treatment of Antibiotic-resistant, Poly-microbial Bone Infection With Bacteriophages and Antibiotics Combination. Clin. Infect Dis. 2019, 69, 2015–2018.
- Eskenazi, A.; Lood, C.; Wubbolts, J.; Hites, M.; Balarjishvili, N.; Leshkasheli, L.; Askilashvili, L.; Kvachadze, L.; van Noort, V.; Wagemans, J.; et al. Combination of pre-adapted bacteriophage therapy and antibiotics for treatment of fracture-related infection due to pandrug-resistant Klebsiella pneumoniae. Nat. Commun. 2022, 13, 302.
- Du, D.; Wang-Kan, X.; Neuberger, A.; van Veen, H.W.; Pos, K.M.; Piddock, L.J.V.; Luisi, B.F. Multidrug efflux pumps: Structure, function and regulation. Nat. Rev. Microbiol. 2018, 16, 523–539.
- Davies, J. Origins and evolution of antibiotic resistance. Microbiologia 1996, 12, 9–16.
- Goldstein, B.P. Resistance to rifampicin: A review. J. Antibiot. 2014, 67, 625–630.
- Ahmad, S.; Mokaddas, E.; Fares, E. Characterization of rpoB mutations in rifampin-resistant clinical Mycobacterium tuberculosis isolates from Kuwait and Dubai. Diagn. Microbiol. Infect. Dis. 2002, 44, 245–252.
- Chen, L.; Gan, X.; Li, N.; Wang, J.; Li, K.; Zhang, H. rpoB gene mutation profile in rifampicin-resistant Mycobacterium tuberculosis clinical isolates from Guizhou, one of the highest incidence rate regions in China. J. Antimicrob. Chemother. 2010, 65, 1299–1301.
- Tan, Y.; Hu, Z.; Zhao, Y.; Cai, X.; Luo, C.; Zou, C.; Liu, X. The beginning of the rpoB gene in addition to the rifampin resistance determination region might be needed for identifying rifampin/rifabutin cross-resistance in multidrug-resistant Mycobacterium tuberculosis isolates from Southern China. J. Clin. Microbiol. 2012, 50, 81–85.
- Siu, G.K.; Zhang, Y.; Lau, T.C.; Lau, R.W.; Ho, P.L.; Yew, W.W.; Tsui, S.K.; Cheng, V.C.; Yuen, K.Y.; Yam, W.C. Mutations outside the rifampicin resistance-determining region associated with rifampicin resistance in Mycobacterium tuberculosis. J. Antimicrob. Chemother. 2011, 66, 730–733.
- Miyachiro, M.M.; Contreras-Martel, C.; Dessen, A. Penicillin-Binding Proteins (PBPs) and Bacterial Cell Wall Elongation Complexes. Subcell. Biochem. 2019, 93, 273–289.
- Miragaia, M. Factors Contributing to the Evolution of mecA-Mediated β-lactam Resistance in Staphylococci: Update and New Insights From Whole Genome Sequencing (WGS). Front. Microbiol. 2018, 9, 2723.
- Nikaido, H. Molecular basis of bacterial outer membrane permeability revisited. Microbiol. Mol. Biol. Rev. 2003, 67, 593–656.
- Chevalier, S.; Bouffartigues, E.; Bodilis, J.; Maillot, O.; Lesouhaitier, O.; Feuilloley, M.G.J.; Orange, N.; Dufour, A.; Cornelis, P. Structure, function and regulation of Pseudomonas aeruginosa porins. FEMS Microbiol. Rev. 2017, 41, 698–722.
- McPhee, J.B.; Tamber, S.; Brazas, M.D.; Lewenza, S.; Hancock, R.E.W. Antibiotic Resistance Due to Reduced Uptake. In Antimicrobial Drug Resistance: Mechanisms of Drug Resistance; Mayers, D.L., Ed.; Humana Press: Totowa, NJ, USA, 2009; pp. 97–110.
- Nadeem, S.F.; Gohar, U.F.; Tahir, S.F.; Mukhtar, H.; Pornpukdeewattana, S.; Nukthamna, P.; Moula Ali, A.M.; Bavisetty, S.C.B.; Massa, S. Antimicrobial resistance: More than 70 years of war between humans and bacteria. Crit. Rev. Microbiol. 2020, 46, 578–599.
- Moya-Torres, A.; Mulvey, M.R.; Kumar, A.; Oresnik, I.J.; Brassinga, A.K.C. The lack of OmpF, but not OmpC, contributes to increased antibiotic resistance in Serratia marcescens. Microbiol. (Read. Engl.) 2014, 160, 1882–1892.
- Ziervogel, B.K.; Roux, B. The Binding of Antibiotics in OmpF Porin. Structure 2013, 21, 76–87.
- Bafna, J.A.; Sans-Serramitjana, E.; Acosta-Gutiérrez, S.; Bodrenko, I.V.; Hörömpöli, D.; Berscheid, A.; Brötz-Oesterhelt, H.; Winterhalter, M.; Ceccarelli, M. Kanamycin Uptake into Escherichia coli Is Facilitated by OmpF and OmpC Porin Channels Located in the Outer Membrane. ACS Infect. Dis. 2020, 6, 1855–1865.
- Tang, X.; Chang, S.; Qiao, W.; Luo, Q.; Chen, Y.; Jia, Z.; Coleman, J.; Zhang, K.; Wang, T.; Zhang, Z.; et al. Structural insights into outer membrane asymmetry maintenance in Gram-negative bacteria by MlaFEDB. Nat. Struct. Mol. Biol. 2021, 28, 81–91.
- Rahman, T.; Yarnall, B.; Doyle, D.A. Efflux drug transporters at the forefront of antimicrobial resistance. Eur. Biophys. J. EBJ 2017, 46, 647–653.
- Amaral, L.; Martins, A.; Spengler, G.; Molnar, J. Efflux pumps of Gram-negative bacteria: What they do, how they do it, with what and how to deal with them. Front. Pharm. 2014, 4, 168.
- Blanco, P.; Hernando-Amado, S.; Reales-Calderon, J.A.; Corona, F.; Lira, F.; Alcalde-Rico, M.; Bernardini, A.; Sanchez, M.B.; Martinez, J.L. Bacterial Multidrug Efflux Pumps: Much More Than Antibiotic Resistance Determinants. Microorganisms 2016, 4, 14.
- Hernando-Amado, S.; Blanco, P.; Alcalde-Rico, M.; Corona, F.; Reales-Calderón, J.A.; Sánchez, M.B.; Martínez, J.L. Multidrug efflux pumps as main players in intrinsic and acquired resistance to antimicrobials. Drug Resist. Updates Rev. Comment. Antimicrob. Anticancer. Chemother. 2016, 28, 13–27.
- Li, X.Z.; Plesiat, P.; Nikaido, H. The challenge of efflux-mediated antibiotic resistance in Gram-negative bacteria. Clin. Microbiol. Rev. 2015, 28, 337–418.
- Kourtesi, C.; Ball, A.R.; Huang, Y.Y.; Jachak, S.M.; Vera, D.M.; Khondkar, P.; Gibbons, S.; Hamblin, M.R.; Tegos, G.P. Microbial efflux systems and inhibitors: Approaches to drug discovery and the challenge of clinical implementation. Open Microbiol. J. 2013, 7, 34–52.
- Costa, S.S.; Viveiros, M.; Amaral, L.; Couto, I. Multidrug Efflux Pumps in Staphylococcus aureus: An Update. Open Microbiol. J. 2013, 7, 59–71.
- Reygaert, W.C. An overview of the antimicrobial resistance mechanisms of bacteria. AIMS Microbiol. 2018, 4, 482–501.
- Swick, M.C.; Morgan-Linnell, S.K.; Carlson, K.M.; Zechiedrich, L. Expression of multidrug efflux pump genes acrAB-tolC, mdfA, and norE in Escherichia coli clinical isolates as a function of fluoroquinolone and multidrug resistance. Antimicrob. Agents Chemother. 2011, 55, 921–924.
- Hansen, L.H.; Jensen, L.B.; Sorensen, H.I.; Sorensen, S.J. Substrate specificity of the OqxAB multidrug resistance pump in Escherichia coli and selected enteric bacteria. J. Antimicrob. Chemother. 2007, 60, 145–147.
- Peterson, E.; Kaur, P. Antibiotic Resistance Mechanisms in Bacteria: Relationships Between Resistance Determinants of Antibiotic Producers, Environmental Bacteria, and Clinical Pathogens. Front. Microbiol. 2018, 9, 2928.
- Tooke, C.L.; Hinchliffe, P.; Bragginton, E.C.; Colenso, C.K.; Hirvonen, V.H.A.; Takebayashi, Y.; Spencer, J. β-Lactamases and β-Lactamase Inhibitors in the 21st Century. J. Mol. Biol. 2019, 431, 3472–3500.
- Ghafourian, S.; Sadeghifard, N.; Soheili, S.; Sekawi, Z. Extended Spectrum Beta-lactamases: Definition, Classification and Epidemiology. Curr. Issues Mol. Biol. 2015, 17, 11–21.
- Sawa, T.; Kooguchi, K.; Moriyama, K. Molecular diversity of extended-spectrum β-lactamases and carbapenemases, and antimicrobial resistance. J. Intensive Care 2020, 8, 13.
- Sköld, O. Aminoglycosides. In Antibiotics and Antibiotic Resistance; John Wiley & Sons: Hoboken, NJ, USA, 2011; Volume 6, pp. 103–113.
- Schwarz, S.; Shen, J.; Kadlec, K.; Wang, Y.; Brenner Michael, G.; Fessler, A.T.; Vester, B. Lincosamides, Streptogramins, Phenicols, and Pleuromutilins: Mode of Action and Mechanisms of Resistance. Cold Spring Harb. Perspect Med. 2016, 6, a027037.
- Lopatkin, A.J.; Bening, S.C.; Manson, A.L.; Stokes, J.M.; Kohanski, M.A.; Badran, A.H.; Earl, A.M.; Cheney, N.J.; Yang, J.H.; Collins, J.J. Clinically relevant mutations in core metabolic genes confer antibiotic resistance. Science 2021, 371, eaba0862.
- Zampieri, M. The genetic underground of antibiotic resistance. Science 2021, 371, 783–784.
- Mee, M.T.; Collins, J.J.; Church, G.M.; Wang, H.H. Syntrophic exchange in synthetic microbial communities. Proc. Natl. Acad. Sci. USA 2014, 111, E2149–E2156.
- Croft, M.T.; Lawrence, A.D.; Raux-Deery, E.; Warren, M.J.; Smith, A.G. Algae acquire vitamin B12 through a symbiotic relationship with bacteria. Nature 2005, 438, 90–93.
- Sieuwerts, S.; Molenaar, D.; van Hijum, S.A.; Beerthuyzen, M.; Stevens, M.J.; Janssen, P.W.; Ingham, C.J.; de Bok, F.A.; de Vos, W.M.; van Hylckama Vlieg, J.E. Mixed-culture transcriptome analysis reveals the molecular basis of mixed-culture growth in Streptococcus thermophilus and Lactobacillus bulgaricus. Appl. Env. Microbiol. 2010, 76, 7775–7784.
- Zelezniak, A.; Andrejev, S.; Ponomarova, O.; Mende, D.R.; Bork, P.; Patil, K.R. Metabolic dependencies drive species co-occurrence in diverse microbial communities. Proc. Natl. Acad. Sci. USA 2015, 112, 6449–6454.
- Seif, Y.; Choudhary, K.S.; Hefner, Y.; Anand, A.; Yang, L.; Palsson, B.O. Metabolic and genetic basis for auxotrophies in Gram-negative species. Proc. Natl. Acad. Sci. USA 2020, 117, 6264–6273.
- Sun, H.; Yao, Z.; Wang, D.; Wu, X.; Lin, Z.; Liu, Y. A deep insight into the toxic mechanism for sulfonamides based on bacterial cell-cell communication. Environ. Int. 2019, 129, 185–193.
- Yu, J.S.L.; Correia-Melo, C.; Zorrilla, F.; Herrera-Dominguez, L.; Wu, M.Y.; Hartl, J.; Campbell, K.; Blasche, S.; Kreidl, M.; Egger, A.S.; et al. Microbial communities form rich extracellular metabolomes that foster metabolic interactions and promote drug tolerance. Nat. Microbiol. 2022, 7, 542–555.
- Wilson, D.N.; Hauryliuk, V.; Atkinson, G.C.; O’Neill, A.J. Target protection as a key antibiotic resistance mechanism. Nat. Rev. Microbiol. 2020, 18, 637–648.
- Sharkey, L.K.; Edwards, T.A.; O’Neill, A.J. ABC-F Proteins Mediate Antibiotic Resistance through Ribosomal Protection. mBio 2016, 7, e01975.
- Sharkey, L.K.R.; O’Neill, A.J. Antibiotic Resistance ABC-F Proteins: Bringing Target Protection into the Limelight. ACS Infect. Dis. 2018, 4, 239–246.
- Su, W.; Kumar, V.; Ding, Y.; Ero, R.; Serra, A.; Lee, B.S.T.; Wong, A.S.W.; Shi, J.; Sze, S.K.; Yang, L.; et al. Ribosome protection by antibiotic resistance ATP-binding cassette protein. Proc. Natl. Acad. Sci. USA 2018, 115, 5157–5162.
- Seo, H.S.; Abedin, S.; Kamp, D.; Wilson, D.N.; Nierhaus, K.H.; Cooperman, B.S. EF-G-dependent GTPase on the ribosome. conformational change and fusidic acid inhibition. Biochemistry 2006, 45, 2504–2514.
- Castanheira, M.; Watters, A.A.; Mendes, R.E.; Farrell, D.J.; Jones, R.N. Occurrence and molecular characterization of fusidic acid resistance mechanisms among Staphylococcus spp. from European countries (2008). J. Antimicrob. Chemother. 2010, 65, 1353–1358.
- McLaws, F.; Chopra, I.; O’Neill, A.J. High prevalence of resistance to fusidic acid in clinical isolates of Staphylococcus epidermidis. J. Antimicrob. Chemother. 2008, 61, 1040–1043.
- George, A.M.; Levy, S.B. Amplifiable resistance to tetracycline, chloramphenicol, and other antibiotics in Escherichia coli: Involvement of a non-plasmid-determined efflux of tetracycline. J. Bacteriol. 1983, 155, 531–540.
- Hao, Z.; Lou, H.; Zhu, R.; Zhu, J.; Zhang, D.; Zhao, B.S.; Zeng, S.; Chen, X.; Chan, J.; He, C.; et al. The multiple antibiotic resistance regulator MarR is a copper sensor in Escherichia coli. Nat. Chem. Biol. 2014, 10, 21–28.
- Ariza, R.R.; Cohen, S.P.; Bachhawat, N.; Levy, S.B.; Demple, B. Repressor mutations in the marRAB operon that activate oxidative stress genes and multiple antibiotic resistance in Escherichia coli. J. Bacteriol. 1994, 176, 143–148.
- McMurry, L.M.; George, A.M.; Levy, S.B. Active efflux of chloramphenicol in susceptible Escherichia coli strains and in multiple-antibiotic-resistant (Mar) mutants. Antimicrob. Agents Chemother. 1994, 38, 542–546.
- Zhang, A.; Rosner, J.L.; Martin, R.G. Transcriptional activation by MarA, SoxS and Rob of two tolC promoters using one binding site: A complex promoter configuration for tolC in Escherichia coli. Mol. Microbiol. 2008, 69, 1450–1455.
- Sharma, P.; Haycocks, J.R.J.; Middlemiss, A.D.; Kettles, R.A.; Sellars, L.E.; Ricci, V.; Piddock, L.J.V.; Grainger, D.C. The multiple antibiotic resistance operon of enteric bacteria controls DNA repair and outer membrane integrity. Nat. Commun. 2017, 8, 1444.
- Banerjee, S.; Lo, K.; Ojkic, N.; Stephens, R.; Scherer, N.F.; Dinner, A.R. Mechanical feedback promotes bacterial adaptation to antibiotics. Nat. Phys. 2021, 17, 403–409.
- Mickiewicz, K.M.; Kawai, Y.; Drage, L.; Gomes, M.C.; Davison, F.; Pickard, R.; Hall, J.; Mostowy, S.; Aldridge, P.D.; Errington, J. Possible role of L-form switching in recurrent urinary tract infection. Nat. Commun. 2019, 10, 4379.
- Tolker-Nielsen, T. Biofilm Development. Microbiol. Spectr. 2015, 3, MB-0001-2014.
- Høiby, N.; Bjarnsholt, T.; Givskov, M.; Molin, S.; Ciofu, O. Antibiotic resistance of bacterial biofilms. Int. J. Antimicrob. Agents 2010, 35, 322–332.
- Bottery, M.J.; Pitchford, J.W.; Friman, V.P. Ecology and evolution of antimicrobial resistance in bacterial communities. ISME J. 2021, 15, 939–948.
- Vega, N.M.; Gore, J. Collective antibiotic resistance: Mechanisms and implications. Curr. Opin. Microbiol. 2014, 21, 28–34.
- Orazi, G.; O’Toole, G.A. Pseudomonas aeruginosa Alters Staphylococcus aureus Sensitivity to Vancomycin in a Biofilm Model of Cystic Fibrosis Infection. mBio 2017, 8, e00873-17.
- Beaudoin, T.; Yau, Y.C.W.; Stapleton, P.J.; Gong, Y.; Wang, P.W.; Guttman, D.S.; Waters, V. Staphylococcus aureus interaction with Pseudomonas aeruginosa biofilm enhances tobramycin resistance. NPJ Biofilms Microbiomes 2017, 3, 25.
- Molina-Santiago, C.; Daddaoua, A.; Fillet, S.; Duque, E.; Ramos, J.L. Interspecies signalling: Pseudomonas putida efflux pump TtgGHI is activated by indole to increase antibiotic resistance. Environ. Microbiol. 2014, 16, 1267–1281.
- Ryan, R.P.; Fouhy, Y.; Garcia, B.F.; Watt, S.A.; Niehaus, K.; Yang, L.; Tolker-Nielsen, T.; Dow, J.M. Interspecies signalling via the Stenotrophomonas maltophilia diffusible signal factor influences biofilm formation and polymyxin tolerance in Pseudomonas aeruginosa. Mol. Microbiol. 2008, 68, 75–86.
- Ling, L.L.; Schneider, T.; Peoples, A.J.; Spoering, A.L.; Engels, I.; Conlon, B.P.; Mueller, A.; Schäberle, T.F.; Hughes, D.E.; Epstein, S.; et al. A new antibiotic kills pathogens without detectable resistance. Nature 2015, 517, 455–459.