Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | M. Faadiel Essop | -- | 2449 | 2022-09-25 15:43:12 |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Teer, E.; Dominick, L.; Mukonowenzou, N.C.; Essop, M.F. Human Immunodeficiency Virus-Related Myocardial Fibrosis. Encyclopedia. Available online: https://encyclopedia.pub/entry/27574 (accessed on 30 June 2026).
Teer E, Dominick L, Mukonowenzou NC, Essop MF. Human Immunodeficiency Virus-Related Myocardial Fibrosis. Encyclopedia. Available at: https://encyclopedia.pub/entry/27574. Accessed June 30, 2026.
Teer, Eman, Leanne Dominick, Nyasha C. Mukonowenzou, M. Faadiel Essop. "Human Immunodeficiency Virus-Related Myocardial Fibrosis" Encyclopedia, https://encyclopedia.pub/entry/27574 (accessed June 30, 2026).
Teer, E., Dominick, L., Mukonowenzou, N.C., & Essop, M.F. (2022, September 26). Human Immunodeficiency Virus-Related Myocardial Fibrosis. In Encyclopedia. https://encyclopedia.pub/entry/27574
Teer, Eman, et al. "Human Immunodeficiency Virus-Related Myocardial Fibrosis." Encyclopedia. Web. 26 September, 2022.
Copy Citation
Chronic inflammation is a characteristic feature of cardiovascular diseases (CVD) and considered a contributor to diastolic dysfunction, heart failure, and sudden cardiac death. This can trigger downstream effects that result in the increased release of pro-coagulant, pro-fibrotic, and pro-inflammatory cytokines. Subsequently, this can lead to an enhanced thrombotic state (by platelet activation), endothelial dysfunction, and myocardial fibrosis. Of note, Studies have revealed that myocardial fibrosis is emerging as a mediator of human immunodeficiency virus (HIV)-related CVD. Together, such factors can eventually result in systolic and diastolic dysfunction, and an increased risk for CVD.
HIV
myocardial fibrosis
platelets
chronic inflammation
sudden cardiac death
heart failure
cardiovascular diseases
1. Introduction
There are currently ~38.4 million human immunodeficiency virus (HIV)-infected individuals globally, with ~28.7 million receiving combination antiretroviral therapy (cART) [1]. Increased access to cART has significantly improved the lifespan of people living with HIV (PLHIV). It has also attenuated viral replication and ensured a relatively well-maintained immune system, together with lowered opportunistic infections and associated mortalities [2]. The main cause of death in PLHIV has therefore shifted from acquired immunodeficiency disease (AIDS)-related immunocompromised states to non-AIDS age-related complications, such as cardiovascular diseases (CVD). In support, the proportion of global deaths due to CVD in PLHIV has increased from 2.5% to 4.6% during the past decade [3][4]. Although HIV-positive patients present with a variety of heart and vascular co-morbidities, certain cardiac disorders manifest with a greater frequency and display geographic variations. For example, in developed countries, PLHIV usually present with metabolic syndrome, hypertension, coronary artery disease, and atherosclerosis [4][5][6]. In contrast, complications such as hypotension, heart failure (HF)/sudden cardiac death (due to HIV-associated cardiomyopathy), and tuberculosis-associated pericarditis are far more prevalent in the sub-Saharan African region [7][8][9][10].
Despite this burgeoning health threat, the underlying mechanisms driving HIV-mediated CVD onset are still being elucidated. Of note, the role of chronic immune activation (despite cART) as a significant mediator in HIV-mediated CVD onset and progression is increasingly being highlighted [11][12]. In agreement, the researchers' laboratory recently found a strong interplay between immune activation, coagulation, and lipid subclass alterations in South African HIV-positive patients [9]. This data also revealed a robust negative correlation between either immune activation or coagulation, and diastolic blood pressure [9]. Chronic inflammation is a characteristic feature of various CVD and is regarded as a key contributor to diastolic dysfunction, HF, and sudden cardiac death [13]. HIV-mediated immune dysregulation can trigger downstream effects that lead to an enhanced release of pro-coagulant, pro-fibrotic, and pro-inflammatory cytokines [14]. This can subsequently result in an increased thrombotic state, endothelial dysfunction, and myocardial fibrosis [15][16]. The culmination of the interplay of such mediators can eventually lead to systolic and diastolic dysfunction and an increased risk for CVD.
While the pathogenesis of HIV-mediated CVD onset and progression is multi-factorial in nature, myocardial fibrosis is emerging as a key mediator underlying the manifestation of systolic and diastolic dysfunction [17]. In support, research findings have revealed prognostic associations between diffuse myocardial fibrosis and left ventricular (LV) remodeling in PLHIV [18][19].
2. HIV Treatment and Cardiovascular Complications
Prior to cART, CVD manifestations of HIV infection included myocardial and peripheral disease, due to the direct effects of HIV, coinfections, and concomitant chronic inflammation [20]. The introduction of cART improved lifespans by viral load reduction and immune system restoration, but also came with side-effects, due to drug-toxicity and metabolic changes (e.g., dyslipidemia, altered glucose handling) [21][22][23]. Furthermore, there are variations between different antiretroviral classes and divergent responses within drug class types [22]. Older generation protein inhibitors, such as lopinavir/ritonavir, and nucleoside reverse transcription inhibitors, such as abacavir, stavudine and zidovudine, can induce dyslipidemia to increase CVD risk [23][24][25][26]. Moreover, body fat distribution changes are still evident years after cessation of antiretroviral use [27][28]. Earlier work, therefore, reported the occurrence of early-onset and aggressive coronary artery disease in PLHIV compared to uninfected individuals [29]. Currently, integrase inhibitors and C-C chemokine receptor 5 antagonists have replaced protease inhibitors as the first line therapy and appear to elicit negligible CVD toxicity, although there are concerns regarding the weight gain associated with their use and hence the need to assess their long-term effects in this context [30].
cART-treated HIV is associated with an increased incidence of myocardial fibrosis [31], as well as both systolic and diastolic LV dysfunction [32], and an up to two-times higher risk of HF [33]. Results from the Veterans Aging Cohort Study [34] showed that this manifests in various forms, such as HF with preserved ejection fraction, borderline HF with preserved ejection fraction, and those with a reduced ejection fraction. Furthermore, the occurrence of such HF subtypes occurs at an earlier stage in the PLHIV population versus uninfected individuals. However, the direct relationship between myocardial inflammation and fibrosis in HIV has been less well studied [13]. For PLHIV who are virally suppressed on cART, the risk of sudden cardiac death levels out to the risk observed in the general population [35]. Moreover, a Taiwanese study found that no specific cART class was associated with increased HF risk [36]. Meanwhile, a relatively small US study on virally suppressed women living with HIV (on integrase strand transfer inhibitors and nucleoside reverse transcription inhibitors) showed increased myocardial fibrosis and lowered diastolic function compared to HIV-negative women [37].
3. Immune Activation and Chronic Inflammation
HIV infection activates the innate and adaptive immune systems, which can result in a state of chronic infection that forms the basis of ongoing immune activation and immunodeficiency [14]. Inflammation is crucial in resolving infections, tissue damage, and maintaining a state of hemostasis [38]. While some degree of immune cell activation is essential to promote suitable responses to injury and activation of tissue repair processes, uncontrolled activation may lead to excess fibrosis and offset its beneficial effects [39].
The innate immune system consists of granulocytes (neutrophils, basophils, eosinophils), mast cells, and antigen presenting cells (macrophages and dendritic cells) [40]. Pathogen-associated molecular patterns and damage-associated molecular patterns can bind to cell surface toll-like receptors, which subsequently results in their activation [14][41]. The activated cells of the innate immune response produce pro-inflammatory cytokines, to further amplify the inflammatory response [38]. The acute inflammatory response starts rapidly, becomes more severe over short periods of time, and usually lasts for a few days [38].
However, if the pathogen-induced stimulation persists, the inflammatory process then acquires new characteristics that are more typically associated with chronic inflammation [38]. This is a slow, long-term state of inflammation that can last for prolonged periods and is induced by cytokines such as interferon-gamma (IFN-γ) that can promote activation of the adaptive immune system [41]. Here, T-cells play a significant role and differentiate into either CD4 (helping to orchestrate immune responses) or CD8 (destroying infected cells) cells [14][41]. Such cells, together with macrophages and natural killer cells, are key players for cell-mediated immunity, while B-cells produce antibodies and are responsible for humoral immunity [40]. Thus, the inflammatory response is the result of a complex interplay between multiple immune cells in the body.
Persistent immune activation and chronic inflammation occur during HIV-infection, despite cART adherence and suppressed viremia [41]. Chronic and persistent CD8+ T-cell activation (the marker of immune activation) rests on three important factors: (1) the persistent detection of HIV-specific effector cytotoxic T cells, (2) the presence of cell surface protein receptors that differentiate naïve T cells into differentiated effector phenotypes [42], and (3) an acute/active cytokine profile detected in serum. Ongoing immune activation and resulting inflammation can lead to immune-related perturbations [43]. Moreover, circulating monocytes and tissue macrophages are both susceptible targets of HIV-1 infection, and the early host response determines whether the infection becomes pathogenic or not. For example, monocytes and macrophages can contribute to the HIV reservoir (and viral persistence) and influence the initiation/extension of immune activation and chronic inflammation, despite cART [44]. Here, the inflammatory response is attenuated (when not required) and becomes chronic if there is a persistent source of activation and/or due to defective control mechanisms [45]. The harmful consequences of persistent immune activation and inflammation during HIV-infection have been extensively reviewed in the previously published literature [13][45][46].
4. Persistent Immune Activation, Chronic Inflammation, and Cardiac Fibrosis
Chronic inflammation and immune dysfunction increase the risk of cardiovascular morbidities and mortalities through endothelial dysfunction, hypercoagulation, and myocardial fibrosis [41][47][48][49][50]. The persistent activation of the innate and adaptive immune systems (monocytes/macrophages and T cells, respectively) results in increased circulating pro-inflammatory and pro-fibrotic cytokines (Figure 1) [12][48][51][52][53][54]. Higher circulating cytokine levels can contribute to hypercoagulation, endothelial dysfunction, and fibrotic remodeling, which increase the risk of CVD onset in PLHIV [31][49][55][56][57][58]. Fibrotic remodeling due to immune dysfunction is an important area of research, due to its detrimental effects on cardiac function, and its links to HF and sudden cardiac death in PLHIV [59]. Myocardial fibrosis is a contributor to sudden cardiac deaths especially in PLHIV that are receiving cART [13][60]. More recently, studies have shown that persistent activation of the innate and adaptive immune responses leads to myocardial fibrosis in PLHIV (Figure 1) [31][58]. Some studies explored subclinical cardiovascular imaging changes using cardiac magnetic resonance and found that HIV-infected patients displayed changes in myocardial function and higher rates of subclinical myocardial inflammation and fibrosis, which worsened with increased severity of the disease [36].
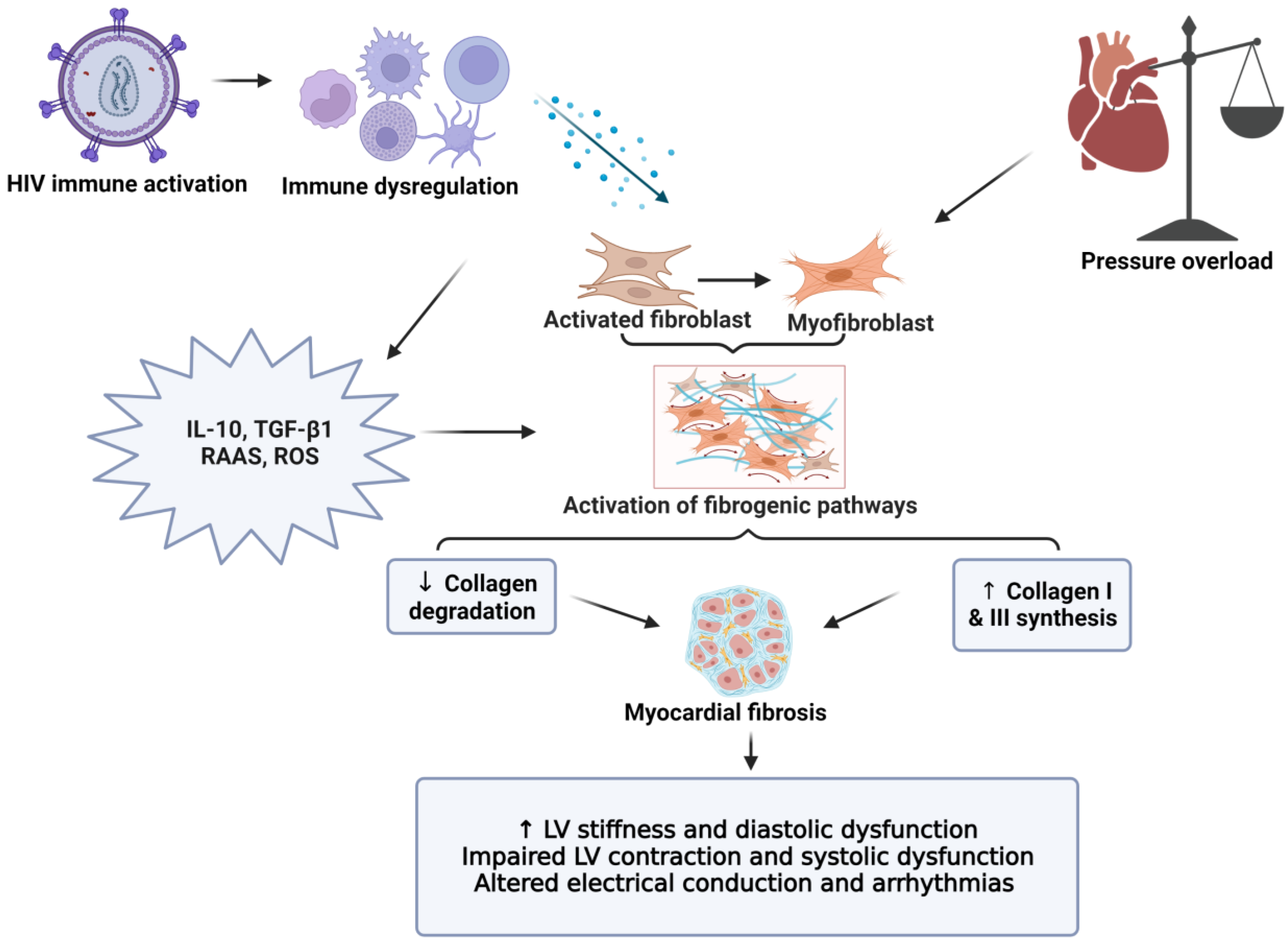
Figure 1. The role of myocardial fibrosis in CVD pathogenesis. The inflammatory hypothesis is considered a main driver of CVD complications in HIV-positive individuals. Persistent immune activation leads to a chronic inflammatory state that includes relatively high levels of inflammatory and pro-fibrotic cytokines (IL-10, TGF-β), together with RAAS activation. This subsequently enhances pro-fibrotic pathways (increased collagen I and III deposition), while also lowering collagen degradation. The increased collagen leads to LV stiffness and diastolic dysfunction (early sign of myocardial fibrosis). Myocardial fibrosis is a contributor to diastolic and systolic dysfunction, HF, and sudden cardiac death. LV: left ventricle, RAAS: renin-angiotensin aldosterone system, ROS: reactive oxygen species, IL: interleukin, and TGF: transforming growth factor.
5. Myocardial Fibrosis: Role in the Pathogenesis of Heart Failure and Sudden Cardiac Death
The modification of the cardiac microenvironment after injury results from the crosstalk between a variety of players such as fibroblasts, endothelial cells, inflammatory and immune cells, soluble factors, and components of the extracellular matrix (ECM) [61]. It is established that cardiac fibrosis is associated with inflammation, exemplified by continuous innate and adaptive immune responses. Myocardial fibrosis is characterized by ECM remodeling, resulting in abnormal matrix composition and leading to impairments in cardiac contractility and function. At first, ECM deposition is defensive and important for wound healing, but unnecessary or prolonged deposition can lead to impairments in tissue function. Fibrosis leads to a stiffer and less compliant heart, eventually contributing to the progression of HF and sudden cardiac death [31].
Of concern, myocardial fibrosis is emerging as a growing cardiac complication in PLHIV. For example, HIV infection (±cART) is linked to an increased incidence of myocardial fibrosis, together with systolic and diastolic LV dysfunction [60][62][63]. Some researchers found that HIV-positive patients exhibited greater evidence of myocardial fibrosis than their negative counterparts, despite relatively normal ejection fractions [31], while others showed a significantly higher prevalence of myocardial fibrosis in PLHIV who suffered mortality due to sudden cardiac deaths [64][65]. Furthermore, a study on HIV-positive patients on cART versus uninfected controls (no CVD history) found that HIV-positive patients displayed a six-fold higher rate of patchy myocardial fibrosis after controlling for age, gender, and coronary artery [60]. In addition, others evaluated associations between HIV serostatus and cardiovascular magnetic resonance imaging and demonstrated that HIV seropositivity was independently associated with greater diffuse non-ischemic fibrosis and a larger left atrial volume [66].
In terms of mechanistic insights, there is some evidence that chronic inflammation can trigger fibrosis, ECM formation, proliferation, and activation of myofibroblasts [13][61]. Activated fibroblasts and myofibroblasts are central effectors in cardiac fibrosis, by functioning as the main source of matrix proteins. Furthermore, the activation of myofibroblasts require the co-operation of growth factors and specialized matrix proteins, which signal through cell surface receptors to activate intracellular signaling pathways that can lead to the synthesis of contractile proteins and the transcription of matrix macromolecules [67]. Several cell types, such as macrophages, mast cells, and lymphocytes (infiltrating the remodeled heart), play an important role in fibroblast activation by secreting a wide range of bioactive mediators, including cytokines such as transforming growth factor (TGF)-β1 and IL-10, and matricellular proteins [61]. Furthermore, the activation of the renin-angiotensin aldosterone system stimulates fibroblast proliferation and ECM protein synthesis in the infarcted and remodeled myocardium, by activation of the angiotensin type 1 receptor or through mineralocorticoid receptor signaling [68]. Although cardiomyocyte death is usually the cause of activation of fibrogenic signals, certain stimuli such as inflammation or pressure overload may activate pro-fibrotic remodeling of the heart [61]. However, despite some progress regarding identification of the underlying mechanisms responsible for the development of myocardial fibrosis during HIV-infection, the associated risk factors and clinical consequences of such pathology still require further elucidation.
Ventricular myocytes are tightly arranged and coupled together, with adjacent layers separated by clefts [61][69][70]. Advanced proteomic methods have revealed that ~90% of the cardiac ECM comprises 10 different proteins, with serum albumin, collagens (collagens I, III, and IV), non-collagenous glycoproteins (fibronectin and laminin), proteoglycans, glucosaminoglycans, and elastins being the most common [71]. The fibrillar collagenous matrix is essentially comprised of type I (>80%) and type III (>10%) collagens [61][70][72]. Fibroblasts regulate collagen turnover by controlling the synthesis and degradation of matrix proteins [73]. As the ECM forms a link between intracellular cytoskeletal proteins and intercellular ones, this allows for the transmission of biochemical signals by mechanosensation [74]. The latter also plays a significant role in activating and differentiating myofibroblasts [74].
There are two types of myocardial fibrosis, namely reactive and replacement. Reactive fibrosis is characterized by excessive extracellular matrix deposition in interstitial or perivascular spaces and is associated with pathological conditions [61]. For example, cardiac structural abnormalities (e.g., HF, arrhythmia, and coronary artery disease) can occur due to the dysregulation of collagen metabolism (synthesis and degradation) [75]. Such structural abnormalities can cause the disruption of myocardial excitation and contraction, thereby leading to impaired systolic and diastolic function (Figure 1) [61][75]. Ventricular dysfunction is the most common cause of HF, including left-sided HF with preserved ejection fraction and reduced ejection fraction with HIV infection [76]. Excessive fibrosis can also cause mechanical stiffness, which may result in the impairment of electric conduction (forming a physical barrier between cardiomyocytes) and lead to impaired cardiac systolic function [61]. Fibrosis can also cause sliding displacement of cardiomyocytes and decrease the number of muscular layers in the ventricular wall, leading to left ventricular dilation [77]. In contrast, replacement fibrosis occurs when there is acute myocardial injury/infarction in the setting of accelerated atherosclerosis associated with HIV. This occurs due to the loss of viable myocardium and results in scar formation and LV remodeling [61][78]. Thus, a balance between replacement and reactive fibrosis is required to prevent cardiac dysfunction [77][78]. As myocardial fibrosis can elicit profound effects on myocardial function and potentially lead to HF and sudden cardiac death, understanding its pathogenesis may help identify promising targets for therapeutic interventions. For example, a recent postmortem study revealed increased rates of sudden cardiac death and myocardial fibrosis in HIV-positive persons versus non-infected individuals [64].
References
- UNAIDS. Global HIV Statistics 2022. Available online: https://www.unaids.org/sites/default/files/media_asset/UNAIDS_FactSheet_en.pdf (accessed on 19 August 2022).
- Hileman, C.O.; Funderburg, N.T. Inflammation, Immune Activation, and Antiretroviral Therapy in HIV. Curr. HIV/AIDS Rep. 2017, 14, 93–100.
- Alonso, A.; Barnes, A.E.; Guest, J.L.; Shah, A.; Shao, I.Y.; Marconi, V. HIV Infection and Incidence of Cardiovascular Diseases: An Analysis of a Large Healthcare Database. J. Am. Heart Assoc. 2019, 8, e012241.
- Feinstein, M.J.; Bogorodskaya, M.; Bloomfield, G.S.; Vedanthan, R.; Siedner, M.J.; Kwan, G.F.; Longenecker, C.T. Cardiovascular Complications of HIV in Endemic Countries. Curr. Cardiol. Rep. 2016, 18, 113.
- Friis-Moller, N.; Ryom, L.; Smith, C.; Weber, R.; Reiss, P.; Dabis, F.; De Wit, S.; Monforte, A.D.; Kirk, O.; Fontas, E.; et al. An updated prediction model of the global risk of cardiovascular disease in HIV-positive persons: The Data-collection on Adverse Effects of Anti-HIV Drugs (D:A:D) study. Eur. J. Prev. Cardiol. 2016, 23, 214–223.
- Gopal, M.; Bhaskaran, A.; Khalife, W.I.; Barbagelata, A. Heart Disease in Patients with HIV/AIDS-An Emerging Clinical Problem. Curr. Cardiol. Rev. 2009, 5, 149–154.
- Bloomfield, G.S.; Hogan, J.W.; Keter, A.; Holland, T.L.; Sang, E.; Kimaiyo, S.; Velazquez, E.J. Blood pressure level impacts risk of death among HIV seropositive adults in Kenya: A retrospective analysis of electronic health records. BMC Infect. Dis. 2014, 14, 284.
- Dominick, L.; Midgley, N.; Swart, L.M.; Sprake, D.; Deshpande, G.; Laher, I.; Joseph, D.; Teer, E.; Essop, M.F. HIV-related cardiovascular diseases: The search for a unifying hypothesis. Am. J. Physiol. Heart Circ. Physiol. 2020, 318, H731–H746.
- Teer, E.; Joseph, D.E.; Driescher, N.; Nell, T.A.; Dominick, L.; Midgley, N.; Deshpande, G.; Page, M.J.; Pretorius, E.; Woudberg, N.J.; et al. HIV and cardiovascular diseases risk: Exploring the interplay between T-cell activation, coagulation, monocyte subsets, and lipid subclass alterations. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H1146–H1157.
- Yuyun, M.F.; Sliwa, K.; Kengne, A.P.; Mocumbi, A.O.; Bukhman, G. Cardiovascular Diseases in Sub-Saharan Africa Compared to High-Income Countries: An Epidemiological Perspective. Glob. Heart 2020, 15, 15.
- Triant, V.A. Cardiovascular disease and HIV infection. Curr. HIV/AIDS Rep. 2013, 10, 199–206.
- Longenecker, C.T.; Sullivan, C.; Baker, J.V. Immune activation and cardiovascular disease in chronic HIV infection. Curr. Opin. HIV AIDS 2016, 11, 216–225.
- Hsue, P.Y.; Tawakol, A. Inflammation and Fibrosis in HIV: Getting to the Heart of the Matter. Circ. Cardiovasc. Imaging 2016, 9, e004427.
- Deeks, S.G.; Tracy, R.; Douek, D.C. Systemic effects of inflammation on health during chronic HIV infection. Immunity 2013, 39, 633–645.
- Musselwhite, L.W.; Sheikh, V.; Norton, T.D.; Rupert, A.; Porter, B.O.; Penzak, S.R.; Skinner, J.; Mican, J.M.; Hadigan, C.; Sereti, I. Markers of endothelial dysfunction, coagulation and tissue fibrosis independently predict venous thromboembolism in HIV. AIDS 2011, 25, 787–795.
- Teer, E.; Essop, M.F. HIV and Cardiovascular Disease: Role of Immunometabolic Perturbations. Physiology 2018, 33, 74–82.
- Hsue, P.Y.; Hunt, P.W.; Ho, J.E.; Farah, H.H.; Schnell, A.; Hoh, R.; Martin, J.N.; Deeks, S.G.; Bolger, A.F. Impact of HIV infection on diastolic function and left ventricular mass. Circ. Heart Fail. 2010, 3, 132–139.
- de Leuw, P.; Arendt, C.T.; Haberl, A.E.; Froadinadl, D.; Kann, G.; Wolf, T.; Stephan, C.; Schuettfort, G.; Vasquez, M.; Arcari, L.; et al. Myocardial Fibrosis and Inflammation by CMR Predict Cardiovascular Outcome in People Living with HIV. JACC Cardiovasc. Imaging 2021, 14, 1548–1557.
- Shuldiner, S.R.; Wong, L.Y.; Peterson, T.E.; Wolfson, J.; Jermy, S.; Saad, H.; Lumbamba, M.A.J.; Singh, A.; Shey, M.; Meintjes, G.; et al. Myocardial Fibrosis Among Antiretroviral Therapy-Treated Persons with Human Immunodeficiency Virus in South Africa. Open Forum Infect. Dis. 2021, 8, ofaa600.
- Acierno, L.J. Cardiac complications in acquired immunodeficiency syndrome (AIDS): A review. J. Am. Coll. Cardiol. 1989, 13, 1144–1154.
- Filardi, P.P.; Paolillo, S.; Marciano, C.; Iorio, A.; Losco, T.; Marsico, F.; Scala, O.; Ruggiero, D.; Ferraro, S.; Chiariello, M. Cardiovascular effects of antiretroviral drugs: Clinical review. Cardiovasc. Hematol. Disord. Drug Targets 2008, 8, 238–244.
- Montessori, V.; Press, N.; Harris, M.; Akagi, L.; Montaner, J.S. Adverse effects of antiretroviral therapy for HIV infection. CMAJ 2004, 170, 229–238.
- Carr, A.; Samaras, K.; Burton, S.; Law, M.; Freund, J.; Chisholm, D.J.; Cooper, D.A. A syndrome of peripheral lipodystrophy, hyperlipidaemia and insulin resistance in patients receiving HIV protease inhibitors. AIDS 1998, 12, F51–F58.
- Matoga, M.M.; Hosseinipour, M.C.; Aga, E.; Ribaudo, H.J.; Kumarasamy, N.; Bartlett, J.; Hughes, M.D.; Team, A.A.S. Hyperlipidaemia in HIV-infected patients on lopinavir/ritonavir monotherapy in resource-limited settings. Antivir. Ther. 2017, 22, 205–213.
- Dorjee, K.; Choden, T.; Baxi, S.M.; Steinmaus, C.; Reingold, A.L. Risk of cardiovascular disease associated with exposure to abacavir among individuals with HIV: A systematic review and meta-analyses of results from 17 epidemiologic studies. Int. J. Antimicrob. Agents 2018, 52, 541–553.
- Abebe, M.; Kinde, S.; Belay, G.; Gebreegziabxier, A.; Challa, F.; Gebeyehu, T.; Nigussie, P.; Tegbaru, B. Antiretroviral treatment associated hyperglycemia and dyslipidemia among HIV infected patients at Burayu Health Center, Addis Ababa, Ethiopia: A cross-sectional comparative study. BMC Res. Notes 2014, 7, 380.
- van Oosterhout, J.J.; Mallewa, J.; Kaunda, S.; Chagoma, N.; Njalale, Y.; Kampira, E.; Mukaka, M.; Heyderman, R.S. Stavudine toxicity in adult longer-term ART patients in Blantyre, Malawi. PLoS ONE 2012, 7, e42029.
- Gelpi, M.; Afzal, S.; Fuchs, A.; Lundgren, J.; Knudsen, A.D.; Drivsholm, N.; Mocroft, A.; Lebech, A.M.; Lindegaard, B.; Kuhl, J.T.; et al. Prior exposure to thymidine analogs and didanosine is associated with long-lasting alterations in adipose tissue distribution and cardiovascular risk factors. AIDS 2019, 33, 675–683.
- Mavroudis, C.A.; Majumder, B.; Loizides, S.; Christophides, T.; Johnson, M.; Rakhit, R.D. Coronary artery disease and HIV; getting to the HAART of the matter. Int. J. Cardiol. 2013, 167, 1147–1153.
- Vos, A.G.; Venter, W.D.F. Cardiovascular toxicity of contemporary antiretroviral therapy. Curr. Opin. HIV AIDS 2021, 16, 286–291.
- Thiara, D.K.; Liu, C.Y.; Raman, F.; Mangat, S.; Purdy, J.B.; Duarte, H.A.; Schmidt, N.; Hur, J.; Sibley, C.T.; Bluemke, D.A.; et al. Abnormal Myocardial Function Is Related to Myocardial Steatosis and Diffuse Myocardial Fibrosis in HIV-Infected Adults. J. Infect. Dis. 2015, 212, 1544–1551.
- Cerrato, E.; D’Ascenzo, F.; Biondi-Zoccai, G.; Calcagno, A.; Frea, S.; Grosso Marra, W.; Castagno, D.; Omede, P.; Quadri, G.; Sciuto, F.; et al. Cardiac dysfunction in pauci symptomatic human immunodeficiency virus patients: A meta-analysis in the highly active antiretroviral therapy era. Eur. Heart J. 2013, 34, 1432–1436.
- Savvoulidis, P.; Butler, J.; Kalogeropoulos, A. Cardiomyopathy and Heart Failure in Patients with HIV Infection. Can. J. Cardiol. 2019, 35, 299–309.
- Freiberg, M.S.; Chang, C.H.; Skanderson, M.; Patterson, O.V.; DuVall, S.L.; Brandt, C.A.; So-Armah, K.A.; Vasan, R.S.; Oursler, K.A.; Gottdiener, J.; et al. Association Between HIV Infection and the Risk of Heart Failure with Reduced Ejection Fraction and Preserved Ejection Fraction in the Antiretroviral Therapy Era: Results From the Veterans Aging Cohort Study. JAMA Cardiol. 2017, 2, 536–546.
- Alvi, R.M.; Neilan, A.M.; Tariq, N.; Hassan, M.O.; Awadalla, M.; Zhang, L.; Afshar, M.; Rokicki, A.; Mulligan, C.P.; Triant, V.A.; et al. The Risk for Sudden Cardiac Death Among Patients Living with Heart Failure and Human Immunodeficiency Virus. JACC Heart Fail 2019, 7, 759–767.
- Yan, C.; Li, R.; Guo, X.; Yu, H.; Li, W.; Li, W.; Ren, M.; Yang, M.; Li, H. Cardiac Involvement in Human Immunodeficiency Virus Infected Patients: An Observational Cardiac Magnetic Resonance Study. Front. Cardiovasc. Med. 2021, 8, 756162.
- Zanni, M.V.; Awadalla, M.; Toribio, M.; Robinson, J.; Stone, L.A.; Cagliero, D.; Rokicki, A.; Mulligan, C.P.; Ho, J.E.; Neilan, A.M.; et al. Immune Correlates of Diffuse Myocardial Fibrosis and Diastolic Dysfunction Among Aging Women with Human Immunodeficiency Virus. J. Infect. Dis. 2020, 221, 1315–1320.
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2018, 9, 7204–7218.
- Lawrence, T.; Gilroy, D.W. Chronic inflammation: A failure of resolution? Int. J. Exp. Pathol. 2007, 88, 85–94.
- Marshall, J.S.; Warrington, R.; Watson, W.; Kim, H.L. An introduction to immunology and immunopathology. Allergy Asthma Clin. Immunol. 2018, 14, 49.
- Pereyra, F.; Lo, J.; Triant, V.A.; Wei, J.; Buzon, M.J.; Fitch, K.V.; Hwang, J.; Campbell, J.H.; Burdo, T.H.; Williams, K.C.; et al. Increased coronary atherosclerosis and immune activation in HIV-1 elite controllers. AIDS 2012, 26, 2409–2412.
- Liu, Z.; Cumberland, W.G.; Hultin, L.E.; Prince, H.E.; Detels, R.; Giorgi, J.V. Elevated CD38 antigen expression on CD8+ T cells is a stronger marker for the risk of chronic HIV disease progression to AIDS and death in the Multicenter AIDS Cohort Study than CD4+ cell count, soluble immune activation markers, or combinations of HLA-DR and CD38 expression. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 1997, 16, 83–92.
- Lohman-Payne, B.; Koster, J.; Gabriel, B.; Chilengi, R.; Forman, L.S.; Heeren, T.; Duffy, C.R.; Herlihy, J.; Crimaldi, S.; Gill, C.; et al. Persistent Immune Activation in Human Immunodeficiency Virus-Infected Pregnant Women Starting Combination Antiretroviral Therapy After Conception. J. Infect. Dis. 2022, 225, 1162–1167.
- Teer, E.; Joseph, D.E.; Glashoff, R.H.; Faadiel Essop, M. Monocyte/Macrophage-Mediated Innate Immunity in HIV-1 Infection: From Early Response to Late Dysregulation and Links to Cardiovascular Diseases Onset. Virol. Sin. 2021, 36, 565–576.
- Appay, V.; Sauce, D. Immune activation and inflammation in HIV-1 infection: Causes and consequences. J. Pathol. 2008, 214, 231–241.
- Paiardini, M.; Muller-Trutwin, M. HIV-associated chronic immune activation. Immunol. Rev. 2013, 254, 78–101.
- Chu, A.J. Tissue factor mediates inflammation. Arch. Biochem. Biophys. 2005, 440, 123–132.
- Sinha, A.; Ma, Y.; Scherzer, R.; Hur, S.; Li, D.; Ganz, P.; Deeks, S.G.; Hsue, P.Y. Role of T-Cell Dysfunction, Inflammation, and Coagulation in Microvascular Disease in HIV. J. Am. Heart Assoc. 2016, 5, e004243.
- Vachiat, A.; McCutcheon, K.; Tsabedze, N.; Zachariah, D.; Manga, P. HIV and Ischemic Heart Disease. J. Am. Coll. Cardiol. 2017, 69, 73–82.
- Witkowski, M.; Landmesser, U.; Rauch, U. Tissue factor as a link between inflammation and coagulation. Trends Cardiovasc. Med. 2016, 26, 297–303.
- Hansson, G.K. Inflammation, atherosclerosis, and coronary artery disease. N. Engl. J. Med. 2005, 352, 1685–1695.
- Mogensen, T.H.; Melchjorsen, J.; Larsen, C.S.; Paludan, S.R. Innate immune recognition and activation during HIV infection. Retrovirology 2010, 7, 54.
- Mooney, S.; Tracy, R.; Osler, T.; Grace, C. Elevated Biomarkers of Inflammation and Coagulation in Patients with HIV Are Associated with Higher Framingham and VACS Risk Index Scores. PLoS ONE 2015, 10, e0144312.
- Serhan, C.N.; Chiang, N.; Van Dyke, T.E. Resolving inflammation: Dual anti-inflammatory and pro-resolution lipid mediators. Nat. Rev. Immunol. 2008, 8, 349–361.
- Arildsen, H.; Sorensen, K.E.; Ingerslev, J.M.; Ostergaard, L.J.; Laursen, A.L. Endothelial dysfunction, increased inflammation, and activated coagulation in HIV-infected patients improve after initiation of highly active antiretroviral therapy. HIV Med. 2013, 14, 1–9.
- Marincowitz, C.; Genis, A.; Goswami, N.; De Boever, P.; Nawrot, T.S.; Strijdom, H. Vascular endothelial dysfunction in the wake of HIV and ART. FEBS J. 2019, 286, 1256–1270.
- Nou, E.; Lo, J.; Grinspoon, S.K. Inflammation, immune activation, and cardiovascular disease in HIV. AIDS 2016, 30, 1495–1509.
- Sager, H.B.; Kessler, T.; Schunkert, H. Monocytes and macrophages in cardiac injury and repair. J. Thorac. Dis. 2017, 9, S30–S35.
- Butler, J.; Kalogeropoulos, A.P.; Anstrom, K.J.; Hsue, P.Y.; Kim, R.J.; Scherzer, R.; Shah, S.J.; Shah, S.H.; Velazquez, E.J.; Hernandez, A.F.; et al. Diastolic Dysfunction in Individuals with Human Immunodeficiency Virus Infection: Literature Review, Rationale and Design of the Characterizing Heart Function on Antiretroviral Therapy (CHART) Study. J. Card. Fail. 2018, 24, 255–265.
- Holloway, C.J.; Ntusi, N.; Suttie, J.; Mahmod, M.; Wainwright, E.; Clutton, G.; Hancock, G.; Beak, P.; Tajar, A.; Piechnik, S.K.; et al. Comprehensive cardiac magnetic resonance imaging and spectroscopy reveal a high burden of myocardial disease in HIV patients. Circulation 2013, 128, 814–822.
- Kong, P.; Christia, P.; Frangogiannis, N.G. The pathogenesis of cardiac fibrosis. Cell. Mol. Life Sci. 2014, 71, 549–574.
- Ahamed, J.; Terry, H.; Choi, M.E.; Laurence, J. Transforming growth factor-β1-mediated cardiac fibrosis: Potential role in HIV and HIV/antiretroviral therapy-linked cardiovascular disease. AIDS 2016, 30, 535–542.
- Utay, N.; Ananworanich, J.; Pinyakorn, S.; Rupert, A.; Sutthichom, D.; Puttamaswin, S. Inflammation persists despite early initiation of ART in acute HIV infection. In Proceedings of the Conference on Retroviruses and Opportunistic Infections, Seattle, DC, USA, 23–26 February 2015; pp. 123–128.
- Tseng, Z.H.; Moffatt, E.; Kim, A.; Vittinghoff, E.; Ursell, P.; Connolly, A.; Olgin, J.E.; Wong, J.K.; Hsue, P.Y. Sudden Cardiac Death and Myocardial Fibrosis, Determined by Autopsy, in Persons with HIV. N. Engl. J. Med. 2021, 384, 2306–2316.
- Tseng, Z.H.; Secemsky, E.A.; Dowdy, D.; Vittinghoff, E.; Moyers, B.; Wong, J.K.; Havlir, D.V.; Hsue, P.Y. Sudden cardiac death in patients with human immunodeficiency virus infection. J. Am. Coll. Cardiol. 2012, 59, 1891–1896.
- Wu, K.C.; Haberlen, S.A.; Plankey, M.W.; Palella, F.J.; Piggott, D.A.; Kirk, G.D.; Margolick, J.B.; Post, W.S. Human immunodeficiency viral infection and differences in interstitial ventricular fibrosis and left atrial size. Eur. Heart J. Cardiovasc. Imaging 2021, 22, 888–895.
- Frangogiannis, N.G. Cardiac fibrosis. Cardiovasc. Res. 2021, 117, 1450–1488.
- Jia, G.; Aroor, A.R.; Hill, M.A.; Sowers, J.R. Role of Renin-Angiotensin-Aldosterone System Activation in Promoting Cardiovascular Fibrosis and Stiffness. Hypertension 2018, 72, 537–548.
- Smaill, B.H.; Zhao, J.; Trew, M.L. Three-dimensional impulse propagation in myocardium: Arrhythmogenic mechanisms at the tissue level. Circ. Res. 2013, 112, 834–848.
- Weber, K.T. Cardiac interstitium in health and disease: The fibrillar collagen network. J. Am. Coll. Cardiol. 1989, 13, 1637–1652.
- Lindsey, M.L.; Jung, M.; Hall, M.E.; DeLeon-Pennell, K.Y. Proteomic analysis of the cardiac extracellular matrix: Clinical research applications. Expert Rev. Proteom. 2018, 15, 105–112.
- Silva, A.C.; Pereira, C.; Fonseca, A.; Pinto-do, O.P.; Nascimento, D.S. Bearing My Heart: The Role of Extracellular Matrix on Cardiac Development, Homeostasis, and Injury Response. Front. Cell Dev. Biol. 2020, 8, 621644.
- Kendall, R.T.; Feghali-Bostwick, C.A. Fibroblasts in fibrosis: Novel roles and mediators. Front. Pharmacol. 2014, 5, 123.
- Liu, T.; Song, D.; Dong, J.; Zhu, P.; Liu, J.; Liu, W.; Ma, X.; Zhao, L.; Ling, S. Current Understanding of the Pathophysiology of Myocardial Fibrosis and Its Quantitative Assessment in Heart Failure. Front. Physiol. 2017, 8, 238.
- Janicki, J.S.; Brower, G.L. The role of myocardial fibrillar collagen in ventricular remodeling and function. J. Card. Fail. 2002, 8, S319–S325.
- Toribio, M.; Neilan, T.G.; Zanni, M.V. Heart failure among people with HIV: Evolving risks, mechanisms, and preventive considerations. Curr. HIV/AIDS Rep. 2019, 16, 371–380.
- Beltrami, C.A.; Finato, N.; Rocco, M.; Feruglio, G.A.; Puricelli, C.; Cigola, E.; Quaini, F.; Sonnenblick, E.H.; Olivetti, G.; Anversa, P. Structural basis of end-stage failure in ischemic cardiomyopathy in humans. Circulation 1994, 89, 151–163.
- Khan, R.; Sheppard, R. Fibrosis in heart disease: Understanding the role of transforming growth factor-β1 in cardiomyopathy, valvular disease and arrhythmia. Immunology 2006, 118, 10–24.
More
Information
Subjects:
Cell Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
596
Revision:
1 time
(View History)
Update Date:
26 Sep 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No