Chronic inflammation is a characteristic feature of cardiovascular diseases (CVD) and considered a contributor to diastolic dysfunction, heart failure, and sudden cardiac death. This can trigger downstream effects that result in the increased release of pro-coagulant, pro-fibrotic, and pro-inflammatory cytokines. Subsequently, this can lead to an enhanced thrombotic state (by platelet activation), endothelial dysfunction, and myocardial fibrosis. Of note, Studies have revealed that myocardial fibrosis is emerging as a mediator of human immunodeficiency virus (HIV)-related CVD. Together, such factors can eventually result in systolic and diastolic dysfunction, and an increased risk for CVD.
- HIV
- myocardial fibrosis
- platelets
- chronic inflammation
- sudden cardiac death
- heart failure
- cardiovascular diseases
1. Introduction
2. HIV Treatment and Cardiovascular Complications
3. Immune Activation and Chronic Inflammation
4. Persistent Immune Activation, Chronic Inflammation, and Cardiac Fibrosis
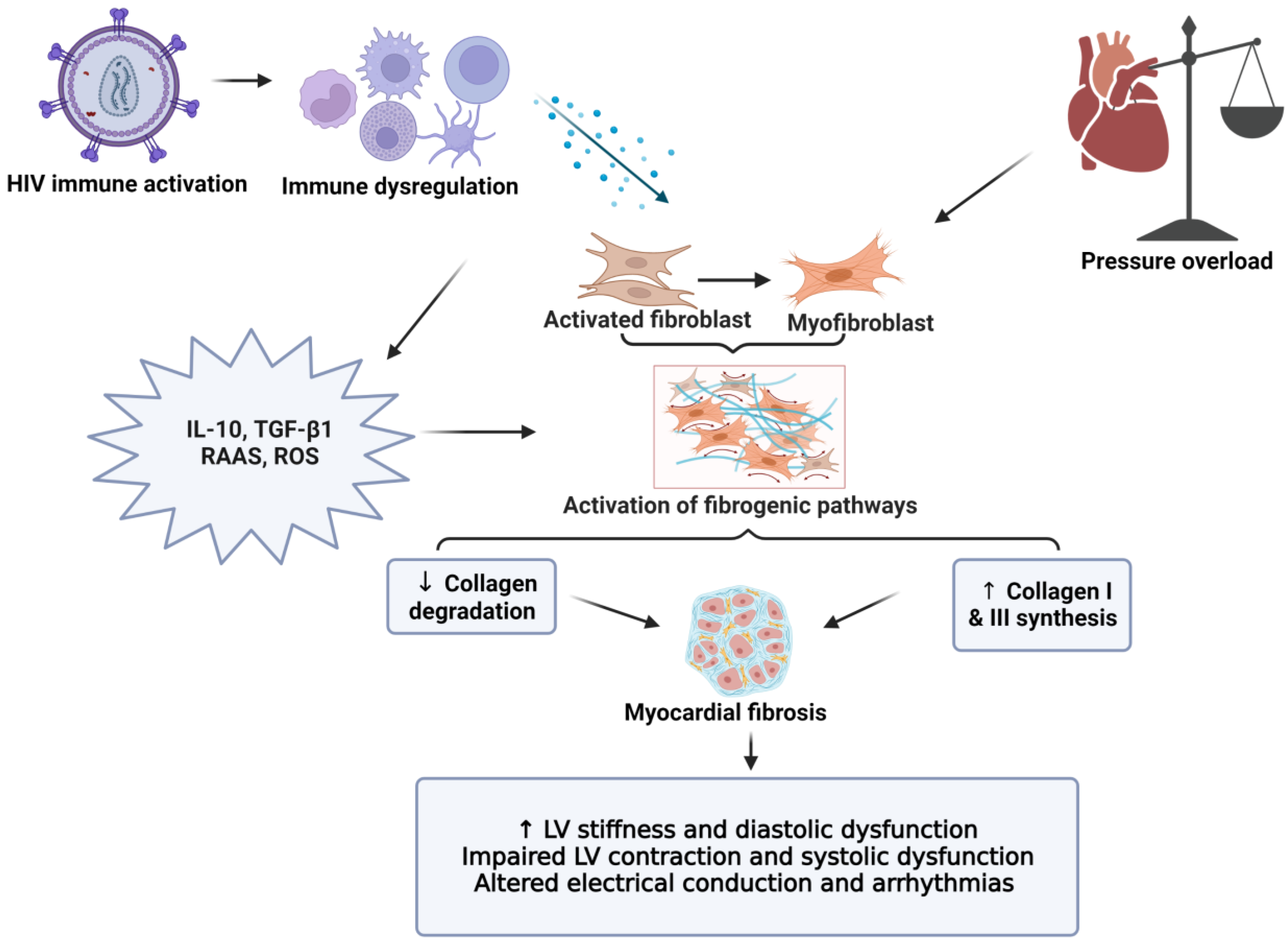
5. Myocardial Fibrosis: Role in the Pathogenesis of Heart Failure and Sudden Cardiac Death
This entry is adapted from the peer-reviewed paper 10.3390/cells11182825
References
- UNAIDS. Global HIV Statistics 2022. Available online: https://www.unaids.org/sites/default/files/media_asset/UNAIDS_FactSheet_en.pdf (accessed on 19 August 2022).
- Hileman, C.O.; Funderburg, N.T. Inflammation, Immune Activation, and Antiretroviral Therapy in HIV. Curr. HIV/AIDS Rep. 2017, 14, 93–100.
- Alonso, A.; Barnes, A.E.; Guest, J.L.; Shah, A.; Shao, I.Y.; Marconi, V. HIV Infection and Incidence of Cardiovascular Diseases: An Analysis of a Large Healthcare Database. J. Am. Heart Assoc. 2019, 8, e012241.
- Feinstein, M.J.; Bogorodskaya, M.; Bloomfield, G.S.; Vedanthan, R.; Siedner, M.J.; Kwan, G.F.; Longenecker, C.T. Cardiovascular Complications of HIV in Endemic Countries. Curr. Cardiol. Rep. 2016, 18, 113.
- Friis-Moller, N.; Ryom, L.; Smith, C.; Weber, R.; Reiss, P.; Dabis, F.; De Wit, S.; Monforte, A.D.; Kirk, O.; Fontas, E.; et al. An updated prediction model of the global risk of cardiovascular disease in HIV-positive persons: The Data-collection on Adverse Effects of Anti-HIV Drugs (D:A:D) study. Eur. J. Prev. Cardiol. 2016, 23, 214–223.
- Gopal, M.; Bhaskaran, A.; Khalife, W.I.; Barbagelata, A. Heart Disease in Patients with HIV/AIDS-An Emerging Clinical Problem. Curr. Cardiol. Rev. 2009, 5, 149–154.
- Bloomfield, G.S.; Hogan, J.W.; Keter, A.; Holland, T.L.; Sang, E.; Kimaiyo, S.; Velazquez, E.J. Blood pressure level impacts risk of death among HIV seropositive adults in Kenya: A retrospective analysis of electronic health records. BMC Infect. Dis. 2014, 14, 284.
- Dominick, L.; Midgley, N.; Swart, L.M.; Sprake, D.; Deshpande, G.; Laher, I.; Joseph, D.; Teer, E.; Essop, M.F. HIV-related cardiovascular diseases: The search for a unifying hypothesis. Am. J. Physiol. Heart Circ. Physiol. 2020, 318, H731–H746.
- Teer, E.; Joseph, D.E.; Driescher, N.; Nell, T.A.; Dominick, L.; Midgley, N.; Deshpande, G.; Page, M.J.; Pretorius, E.; Woudberg, N.J.; et al. HIV and cardiovascular diseases risk: Exploring the interplay between T-cell activation, coagulation, monocyte subsets, and lipid subclass alterations. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H1146–H1157.
- Yuyun, M.F.; Sliwa, K.; Kengne, A.P.; Mocumbi, A.O.; Bukhman, G. Cardiovascular Diseases in Sub-Saharan Africa Compared to High-Income Countries: An Epidemiological Perspective. Glob. Heart 2020, 15, 15.
- Triant, V.A. Cardiovascular disease and HIV infection. Curr. HIV/AIDS Rep. 2013, 10, 199–206.
- Longenecker, C.T.; Sullivan, C.; Baker, J.V. Immune activation and cardiovascular disease in chronic HIV infection. Curr. Opin. HIV AIDS 2016, 11, 216–225.
- Hsue, P.Y.; Tawakol, A. Inflammation and Fibrosis in HIV: Getting to the Heart of the Matter. Circ. Cardiovasc. Imaging 2016, 9, e004427.
- Deeks, S.G.; Tracy, R.; Douek, D.C. Systemic effects of inflammation on health during chronic HIV infection. Immunity 2013, 39, 633–645.
- Musselwhite, L.W.; Sheikh, V.; Norton, T.D.; Rupert, A.; Porter, B.O.; Penzak, S.R.; Skinner, J.; Mican, J.M.; Hadigan, C.; Sereti, I. Markers of endothelial dysfunction, coagulation and tissue fibrosis independently predict venous thromboembolism in HIV. AIDS 2011, 25, 787–795.
- Teer, E.; Essop, M.F. HIV and Cardiovascular Disease: Role of Immunometabolic Perturbations. Physiology 2018, 33, 74–82.
- Hsue, P.Y.; Hunt, P.W.; Ho, J.E.; Farah, H.H.; Schnell, A.; Hoh, R.; Martin, J.N.; Deeks, S.G.; Bolger, A.F. Impact of HIV infection on diastolic function and left ventricular mass. Circ. Heart Fail. 2010, 3, 132–139.
- de Leuw, P.; Arendt, C.T.; Haberl, A.E.; Froadinadl, D.; Kann, G.; Wolf, T.; Stephan, C.; Schuettfort, G.; Vasquez, M.; Arcari, L.; et al. Myocardial Fibrosis and Inflammation by CMR Predict Cardiovascular Outcome in People Living with HIV. JACC Cardiovasc. Imaging 2021, 14, 1548–1557.
- Shuldiner, S.R.; Wong, L.Y.; Peterson, T.E.; Wolfson, J.; Jermy, S.; Saad, H.; Lumbamba, M.A.J.; Singh, A.; Shey, M.; Meintjes, G.; et al. Myocardial Fibrosis Among Antiretroviral Therapy-Treated Persons with Human Immunodeficiency Virus in South Africa. Open Forum Infect. Dis. 2021, 8, ofaa600.
- Acierno, L.J. Cardiac complications in acquired immunodeficiency syndrome (AIDS): A review. J. Am. Coll. Cardiol. 1989, 13, 1144–1154.
- Filardi, P.P.; Paolillo, S.; Marciano, C.; Iorio, A.; Losco, T.; Marsico, F.; Scala, O.; Ruggiero, D.; Ferraro, S.; Chiariello, M. Cardiovascular effects of antiretroviral drugs: Clinical review. Cardiovasc. Hematol. Disord. Drug Targets 2008, 8, 238–244.
- Montessori, V.; Press, N.; Harris, M.; Akagi, L.; Montaner, J.S. Adverse effects of antiretroviral therapy for HIV infection. CMAJ 2004, 170, 229–238.
- Carr, A.; Samaras, K.; Burton, S.; Law, M.; Freund, J.; Chisholm, D.J.; Cooper, D.A. A syndrome of peripheral lipodystrophy, hyperlipidaemia and insulin resistance in patients receiving HIV protease inhibitors. AIDS 1998, 12, F51–F58.
- Matoga, M.M.; Hosseinipour, M.C.; Aga, E.; Ribaudo, H.J.; Kumarasamy, N.; Bartlett, J.; Hughes, M.D.; Team, A.A.S. Hyperlipidaemia in HIV-infected patients on lopinavir/ritonavir monotherapy in resource-limited settings. Antivir. Ther. 2017, 22, 205–213.
- Dorjee, K.; Choden, T.; Baxi, S.M.; Steinmaus, C.; Reingold, A.L. Risk of cardiovascular disease associated with exposure to abacavir among individuals with HIV: A systematic review and meta-analyses of results from 17 epidemiologic studies. Int. J. Antimicrob. Agents 2018, 52, 541–553.
- Abebe, M.; Kinde, S.; Belay, G.; Gebreegziabxier, A.; Challa, F.; Gebeyehu, T.; Nigussie, P.; Tegbaru, B. Antiretroviral treatment associated hyperglycemia and dyslipidemia among HIV infected patients at Burayu Health Center, Addis Ababa, Ethiopia: A cross-sectional comparative study. BMC Res. Notes 2014, 7, 380.
- van Oosterhout, J.J.; Mallewa, J.; Kaunda, S.; Chagoma, N.; Njalale, Y.; Kampira, E.; Mukaka, M.; Heyderman, R.S. Stavudine toxicity in adult longer-term ART patients in Blantyre, Malawi. PLoS ONE 2012, 7, e42029.
- Gelpi, M.; Afzal, S.; Fuchs, A.; Lundgren, J.; Knudsen, A.D.; Drivsholm, N.; Mocroft, A.; Lebech, A.M.; Lindegaard, B.; Kuhl, J.T.; et al. Prior exposure to thymidine analogs and didanosine is associated with long-lasting alterations in adipose tissue distribution and cardiovascular risk factors. AIDS 2019, 33, 675–683.
- Mavroudis, C.A.; Majumder, B.; Loizides, S.; Christophides, T.; Johnson, M.; Rakhit, R.D. Coronary artery disease and HIV; getting to the HAART of the matter. Int. J. Cardiol. 2013, 167, 1147–1153.
- Vos, A.G.; Venter, W.D.F. Cardiovascular toxicity of contemporary antiretroviral therapy. Curr. Opin. HIV AIDS 2021, 16, 286–291.
- Thiara, D.K.; Liu, C.Y.; Raman, F.; Mangat, S.; Purdy, J.B.; Duarte, H.A.; Schmidt, N.; Hur, J.; Sibley, C.T.; Bluemke, D.A.; et al. Abnormal Myocardial Function Is Related to Myocardial Steatosis and Diffuse Myocardial Fibrosis in HIV-Infected Adults. J. Infect. Dis. 2015, 212, 1544–1551.
- Cerrato, E.; D’Ascenzo, F.; Biondi-Zoccai, G.; Calcagno, A.; Frea, S.; Grosso Marra, W.; Castagno, D.; Omede, P.; Quadri, G.; Sciuto, F.; et al. Cardiac dysfunction in pauci symptomatic human immunodeficiency virus patients: A meta-analysis in the highly active antiretroviral therapy era. Eur. Heart J. 2013, 34, 1432–1436.
- Savvoulidis, P.; Butler, J.; Kalogeropoulos, A. Cardiomyopathy and Heart Failure in Patients with HIV Infection. Can. J. Cardiol. 2019, 35, 299–309.
- Freiberg, M.S.; Chang, C.H.; Skanderson, M.; Patterson, O.V.; DuVall, S.L.; Brandt, C.A.; So-Armah, K.A.; Vasan, R.S.; Oursler, K.A.; Gottdiener, J.; et al. Association Between HIV Infection and the Risk of Heart Failure with Reduced Ejection Fraction and Preserved Ejection Fraction in the Antiretroviral Therapy Era: Results From the Veterans Aging Cohort Study. JAMA Cardiol. 2017, 2, 536–546.
- Alvi, R.M.; Neilan, A.M.; Tariq, N.; Hassan, M.O.; Awadalla, M.; Zhang, L.; Afshar, M.; Rokicki, A.; Mulligan, C.P.; Triant, V.A.; et al. The Risk for Sudden Cardiac Death Among Patients Living with Heart Failure and Human Immunodeficiency Virus. JACC Heart Fail 2019, 7, 759–767.
- Yan, C.; Li, R.; Guo, X.; Yu, H.; Li, W.; Li, W.; Ren, M.; Yang, M.; Li, H. Cardiac Involvement in Human Immunodeficiency Virus Infected Patients: An Observational Cardiac Magnetic Resonance Study. Front. Cardiovasc. Med. 2021, 8, 756162.
- Zanni, M.V.; Awadalla, M.; Toribio, M.; Robinson, J.; Stone, L.A.; Cagliero, D.; Rokicki, A.; Mulligan, C.P.; Ho, J.E.; Neilan, A.M.; et al. Immune Correlates of Diffuse Myocardial Fibrosis and Diastolic Dysfunction Among Aging Women with Human Immunodeficiency Virus. J. Infect. Dis. 2020, 221, 1315–1320.
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2018, 9, 7204–7218.
- Lawrence, T.; Gilroy, D.W. Chronic inflammation: A failure of resolution? Int. J. Exp. Pathol. 2007, 88, 85–94.
- Marshall, J.S.; Warrington, R.; Watson, W.; Kim, H.L. An introduction to immunology and immunopathology. Allergy Asthma Clin. Immunol. 2018, 14, 49.
- Pereyra, F.; Lo, J.; Triant, V.A.; Wei, J.; Buzon, M.J.; Fitch, K.V.; Hwang, J.; Campbell, J.H.; Burdo, T.H.; Williams, K.C.; et al. Increased coronary atherosclerosis and immune activation in HIV-1 elite controllers. AIDS 2012, 26, 2409–2412.
- Liu, Z.; Cumberland, W.G.; Hultin, L.E.; Prince, H.E.; Detels, R.; Giorgi, J.V. Elevated CD38 antigen expression on CD8+ T cells is a stronger marker for the risk of chronic HIV disease progression to AIDS and death in the Multicenter AIDS Cohort Study than CD4+ cell count, soluble immune activation markers, or combinations of HLA-DR and CD38 expression. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 1997, 16, 83–92.
- Lohman-Payne, B.; Koster, J.; Gabriel, B.; Chilengi, R.; Forman, L.S.; Heeren, T.; Duffy, C.R.; Herlihy, J.; Crimaldi, S.; Gill, C.; et al. Persistent Immune Activation in Human Immunodeficiency Virus-Infected Pregnant Women Starting Combination Antiretroviral Therapy After Conception. J. Infect. Dis. 2022, 225, 1162–1167.
- Teer, E.; Joseph, D.E.; Glashoff, R.H.; Faadiel Essop, M. Monocyte/Macrophage-Mediated Innate Immunity in HIV-1 Infection: From Early Response to Late Dysregulation and Links to Cardiovascular Diseases Onset. Virol. Sin. 2021, 36, 565–576.
- Appay, V.; Sauce, D. Immune activation and inflammation in HIV-1 infection: Causes and consequences. J. Pathol. 2008, 214, 231–241.
- Paiardini, M.; Muller-Trutwin, M. HIV-associated chronic immune activation. Immunol. Rev. 2013, 254, 78–101.
- Chu, A.J. Tissue factor mediates inflammation. Arch. Biochem. Biophys. 2005, 440, 123–132.
- Sinha, A.; Ma, Y.; Scherzer, R.; Hur, S.; Li, D.; Ganz, P.; Deeks, S.G.; Hsue, P.Y. Role of T-Cell Dysfunction, Inflammation, and Coagulation in Microvascular Disease in HIV. J. Am. Heart Assoc. 2016, 5, e004243.
- Vachiat, A.; McCutcheon, K.; Tsabedze, N.; Zachariah, D.; Manga, P. HIV and Ischemic Heart Disease. J. Am. Coll. Cardiol. 2017, 69, 73–82.
- Witkowski, M.; Landmesser, U.; Rauch, U. Tissue factor as a link between inflammation and coagulation. Trends Cardiovasc. Med. 2016, 26, 297–303.
- Hansson, G.K. Inflammation, atherosclerosis, and coronary artery disease. N. Engl. J. Med. 2005, 352, 1685–1695.
- Mogensen, T.H.; Melchjorsen, J.; Larsen, C.S.; Paludan, S.R. Innate immune recognition and activation during HIV infection. Retrovirology 2010, 7, 54.
- Mooney, S.; Tracy, R.; Osler, T.; Grace, C. Elevated Biomarkers of Inflammation and Coagulation in Patients with HIV Are Associated with Higher Framingham and VACS Risk Index Scores. PLoS ONE 2015, 10, e0144312.
- Serhan, C.N.; Chiang, N.; Van Dyke, T.E. Resolving inflammation: Dual anti-inflammatory and pro-resolution lipid mediators. Nat. Rev. Immunol. 2008, 8, 349–361.
- Arildsen, H.; Sorensen, K.E.; Ingerslev, J.M.; Ostergaard, L.J.; Laursen, A.L. Endothelial dysfunction, increased inflammation, and activated coagulation in HIV-infected patients improve after initiation of highly active antiretroviral therapy. HIV Med. 2013, 14, 1–9.
- Marincowitz, C.; Genis, A.; Goswami, N.; De Boever, P.; Nawrot, T.S.; Strijdom, H. Vascular endothelial dysfunction in the wake of HIV and ART. FEBS J. 2019, 286, 1256–1270.
- Nou, E.; Lo, J.; Grinspoon, S.K. Inflammation, immune activation, and cardiovascular disease in HIV. AIDS 2016, 30, 1495–1509.
- Sager, H.B.; Kessler, T.; Schunkert, H. Monocytes and macrophages in cardiac injury and repair. J. Thorac. Dis. 2017, 9, S30–S35.
- Butler, J.; Kalogeropoulos, A.P.; Anstrom, K.J.; Hsue, P.Y.; Kim, R.J.; Scherzer, R.; Shah, S.J.; Shah, S.H.; Velazquez, E.J.; Hernandez, A.F.; et al. Diastolic Dysfunction in Individuals with Human Immunodeficiency Virus Infection: Literature Review, Rationale and Design of the Characterizing Heart Function on Antiretroviral Therapy (CHART) Study. J. Card. Fail. 2018, 24, 255–265.
- Holloway, C.J.; Ntusi, N.; Suttie, J.; Mahmod, M.; Wainwright, E.; Clutton, G.; Hancock, G.; Beak, P.; Tajar, A.; Piechnik, S.K.; et al. Comprehensive cardiac magnetic resonance imaging and spectroscopy reveal a high burden of myocardial disease in HIV patients. Circulation 2013, 128, 814–822.
- Kong, P.; Christia, P.; Frangogiannis, N.G. The pathogenesis of cardiac fibrosis. Cell. Mol. Life Sci. 2014, 71, 549–574.
- Ahamed, J.; Terry, H.; Choi, M.E.; Laurence, J. Transforming growth factor-β1-mediated cardiac fibrosis: Potential role in HIV and HIV/antiretroviral therapy-linked cardiovascular disease. AIDS 2016, 30, 535–542.
- Utay, N.; Ananworanich, J.; Pinyakorn, S.; Rupert, A.; Sutthichom, D.; Puttamaswin, S. Inflammation persists despite early initiation of ART in acute HIV infection. In Proceedings of the Conference on Retroviruses and Opportunistic Infections, Seattle, DC, USA, 23–26 February 2015; pp. 123–128.
- Tseng, Z.H.; Moffatt, E.; Kim, A.; Vittinghoff, E.; Ursell, P.; Connolly, A.; Olgin, J.E.; Wong, J.K.; Hsue, P.Y. Sudden Cardiac Death and Myocardial Fibrosis, Determined by Autopsy, in Persons with HIV. N. Engl. J. Med. 2021, 384, 2306–2316.
- Tseng, Z.H.; Secemsky, E.A.; Dowdy, D.; Vittinghoff, E.; Moyers, B.; Wong, J.K.; Havlir, D.V.; Hsue, P.Y. Sudden cardiac death in patients with human immunodeficiency virus infection. J. Am. Coll. Cardiol. 2012, 59, 1891–1896.
- Wu, K.C.; Haberlen, S.A.; Plankey, M.W.; Palella, F.J.; Piggott, D.A.; Kirk, G.D.; Margolick, J.B.; Post, W.S. Human immunodeficiency viral infection and differences in interstitial ventricular fibrosis and left atrial size. Eur. Heart J. Cardiovasc. Imaging 2021, 22, 888–895.
- Frangogiannis, N.G. Cardiac fibrosis. Cardiovasc. Res. 2021, 117, 1450–1488.
- Jia, G.; Aroor, A.R.; Hill, M.A.; Sowers, J.R. Role of Renin-Angiotensin-Aldosterone System Activation in Promoting Cardiovascular Fibrosis and Stiffness. Hypertension 2018, 72, 537–548.
- Smaill, B.H.; Zhao, J.; Trew, M.L. Three-dimensional impulse propagation in myocardium: Arrhythmogenic mechanisms at the tissue level. Circ. Res. 2013, 112, 834–848.
- Weber, K.T. Cardiac interstitium in health and disease: The fibrillar collagen network. J. Am. Coll. Cardiol. 1989, 13, 1637–1652.
- Lindsey, M.L.; Jung, M.; Hall, M.E.; DeLeon-Pennell, K.Y. Proteomic analysis of the cardiac extracellular matrix: Clinical research applications. Expert Rev. Proteom. 2018, 15, 105–112.
- Silva, A.C.; Pereira, C.; Fonseca, A.; Pinto-do, O.P.; Nascimento, D.S. Bearing My Heart: The Role of Extracellular Matrix on Cardiac Development, Homeostasis, and Injury Response. Front. Cell Dev. Biol. 2020, 8, 621644.
- Kendall, R.T.; Feghali-Bostwick, C.A. Fibroblasts in fibrosis: Novel roles and mediators. Front. Pharmacol. 2014, 5, 123.
- Liu, T.; Song, D.; Dong, J.; Zhu, P.; Liu, J.; Liu, W.; Ma, X.; Zhao, L.; Ling, S. Current Understanding of the Pathophysiology of Myocardial Fibrosis and Its Quantitative Assessment in Heart Failure. Front. Physiol. 2017, 8, 238.
- Janicki, J.S.; Brower, G.L. The role of myocardial fibrillar collagen in ventricular remodeling and function. J. Card. Fail. 2002, 8, S319–S325.
- Toribio, M.; Neilan, T.G.; Zanni, M.V. Heart failure among people with HIV: Evolving risks, mechanisms, and preventive considerations. Curr. HIV/AIDS Rep. 2019, 16, 371–380.
- Beltrami, C.A.; Finato, N.; Rocco, M.; Feruglio, G.A.; Puricelli, C.; Cigola, E.; Quaini, F.; Sonnenblick, E.H.; Olivetti, G.; Anversa, P. Structural basis of end-stage failure in ischemic cardiomyopathy in humans. Circulation 1994, 89, 151–163.
- Khan, R.; Sheppard, R. Fibrosis in heart disease: Understanding the role of transforming growth factor-β1 in cardiomyopathy, valvular disease and arrhythmia. Immunology 2006, 118, 10–24.