Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Sangeeta Ttiwari | -- | 2031 | 2022-09-22 18:51:14 | | | |
| 2 | Rita Xu | Meta information modification | 2031 | 2022-09-23 03:09:39 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Togre, N.S.; Vargas, A.M.; Bhargavi, G.; Mallakuntla, M.K.; Tiwari, S. Fragment-Based Drug Discovery against Mycobacteria. Encyclopedia. Available online: https://encyclopedia.pub/entry/27497 (accessed on 08 August 2026).
Togre NS, Vargas AM, Bhargavi G, Mallakuntla MK, Tiwari S. Fragment-Based Drug Discovery against Mycobacteria. Encyclopedia. Available at: https://encyclopedia.pub/entry/27497. Accessed August 08, 2026.
Togre, Namdev S., Ana M. Vargas, Gunapati Bhargavi, Mohan Krishna Mallakuntla, Sangeeta Tiwari. "Fragment-Based Drug Discovery against Mycobacteria" Encyclopedia, https://encyclopedia.pub/entry/27497 (accessed August 08, 2026).
Togre, N.S., Vargas, A.M., Bhargavi, G., Mallakuntla, M.K., & Tiwari, S. (2022, September 22). Fragment-Based Drug Discovery against Mycobacteria. In Encyclopedia. https://encyclopedia.pub/entry/27497
Togre, Namdev S., et al. "Fragment-Based Drug Discovery against Mycobacteria." Encyclopedia. Web. 22 September, 2022.
Copy Citation
The emergence of drug-resistant mycobacteria, including Mycobacterium tuberculosis (Mtb) and non-tuberculous mycobacteria (NTM), poses an increasing global threat that urgently demands the development of new potent anti-mycobacterial drugs. One of the approaches toward the identification of new drugs is fragment-based drug discovery (FBDD), which is the most ingenious among other drug discovery models, such as structure-based drug design (SBDD) and high-throughput screening. Specialized techniques, such as X-ray crystallography, nuclear magnetic resonance spectroscopy, and many others, are part of the drug discovery approach to combat the Mtb and NTM global menaces.
NTM
mycobacteria
FBDD
drug discovery
NTM drug discovery
1. Introduction
The World Health Organization (WHO) has reported global tuberculosis (TB) with an estimated 10 million active new cases, 1.5 million deaths, and 1.7 billion latent infections in 2020 [1]. Although the effective treatment of TB has been available for over four decades, the occurrence of drug resistance (DR) and multi-drug resistance (MDR) is a major challenge during the treatment course. According to the recent WHO report, although the total number of DR cases decreased by 22% in 2019–2020, the existing drug regimens are ineffective in combating the disease progression [1][2]. Recent studies identified fragment-based drug discovery (FBDD) as a key approach in the field of drug discovery to find new targets through high-throughput screening (HTS) techniques [3][4]. The identification of even smaller molecules, the druggability of biological targets, and alternate inhibition sites on established drugs are key to the FBDD’s success [5][6]. Based on this approach, more than 40 new compounds have been identified and are currently in clinical trials [7][8][9]. Figure 1 shows the basic approach used to identify new compounds in FBDD.
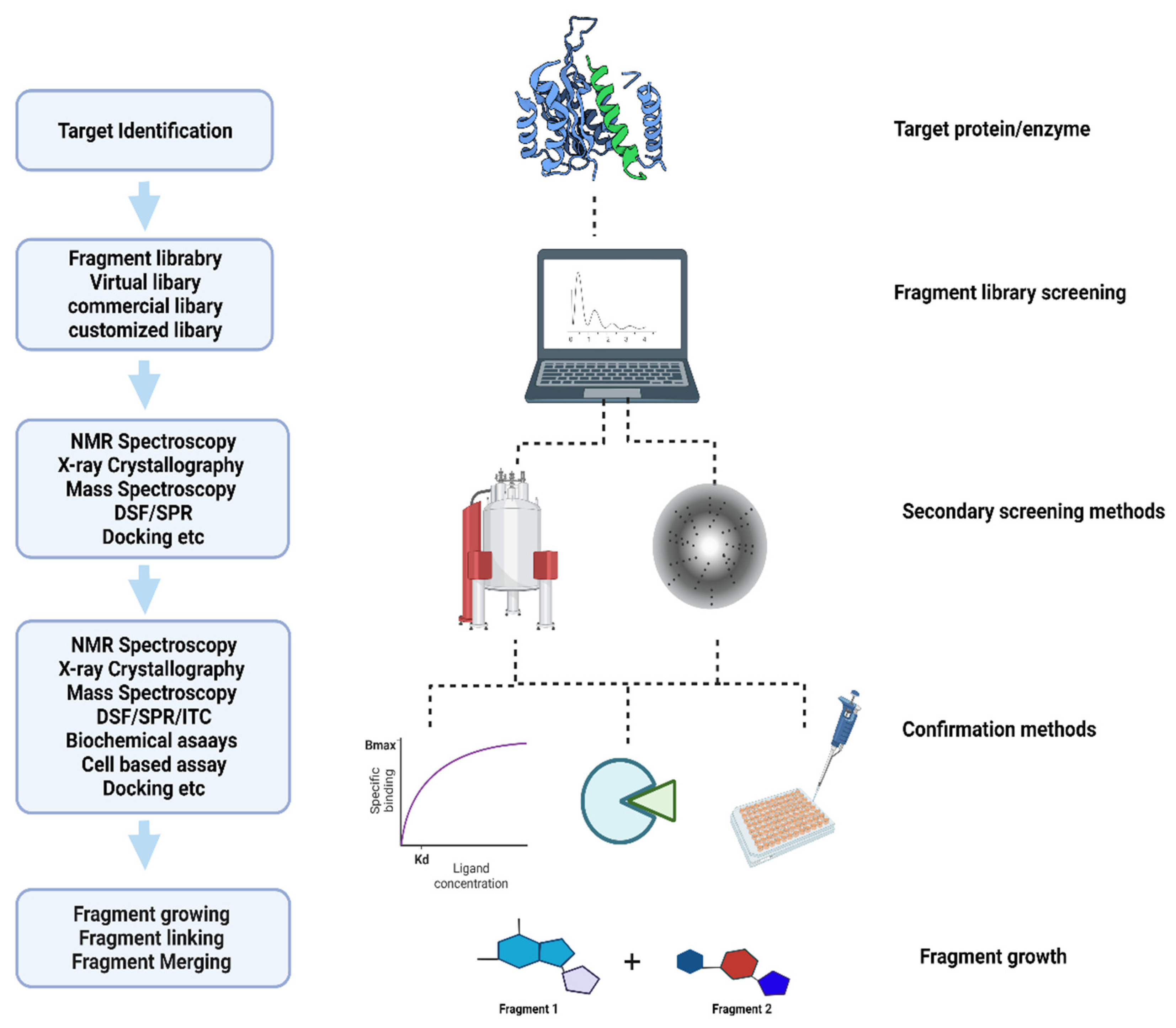
Figure 1. Flow chart of fragment-based drug discovery.
This FBDD approach has evolved steadily over the last decade. The application of fragment library screening has been successfully applied to a wide range of infectious diseases, including, but not limited to, Leishmania, flaviviruses, non-tuberculosis mycobacteria (NTM), and Mtb [10]. The discovery of the phosphoribosylaminoimidazole succinocarboxamide synthetase enzyme (PurC) as a potential inhibitor target in both M. abscessus and Mtb is also a significant breakthrough in FBDD [11]. It is crucial to understand how beneficial the FBDD approach is in developing new molecules with possible potential therapeutic activity against NTM and Mtb infections.
2. Challenges in Fragment-Based Drug Discovery
Drug discovery for NTMs and MTB has been successfully accomplished using the FBDD. This strategy, however, faces a number of obstacles and challenges with its methodologies. Researchers have covered a lot of them in this area, which will help to overcome the challenges listed below and pave the way for successful drug discovery (Table 1).
Table 1. Challenges in the FBDD approach.
| Method/Molecules | Challenges |
|---|---|
| X-ray crystallography |
|
| X-ray crystal structures |
|
| Crystal soaking |
|
| Molecular docking with smaller fragments |
|
| Homology modeling |
|
| Stoichiometric approach |
|
| Fragment screening |
|
| Fragments |
|
| Low efficiency fragments |
|
| Fragment potential hits |
|
2.1. Specialized Methods Are Needed to Detect Fragment Binding in Libraries
The fragments form high-attribute interactions if they have suitable ligand efficiencies (LE), which are ineffective due to their smaller size and weak affinity [12]. It is well known that selected fragment libraries follow the “rule of three”. For example, using three-dimensional fragments instead of flat structures, identifying differences at fragment binding sites and protein binding sites, and determining the potent target hits for fragment protein interaction. Although there are arguments against using the rule of three, it is considered the preferred model for fragment selection in libraries [13][14][15]. Various approaches, such as NMR, surface plasmon resonance (SPR), and X-ray screening, have greater sensitivity and are specifically used to screen larger fragments in higher libraries in fragment-based drug discovery (FBDD) [16]. Additionally, thermal denaturation, thermal electrophoresis, capillary electrophoresis, mass spectrometry, and isothermal titration calorimetry have also been widely used in FBDD despite their limitations.
2.2. Optimization of Fragment Hits Using Computational Tools
Computational tools, such as de novo design, combinatorial docking, and interactive optimization, and their approaches, play a key role in the optimization process through fragment linking that eventually reduces the time required for increasing the efficacy of potent fragment leads [17][18]. Optimization and proper knowledge of specific fragments is a pre-requisite for FBDD [19]. The large size of targeted molecules or the existence of targets as multiprotein complexes significantly impacts the generation of robust crystals, leading to the hindrance of using the FBDD approach for screening fragments [8]. The optimization of fragment hits also depends on detection techniques considered for FBDD; the correlation of fragment hits must be carefully considered. For example, 5% of fragment interaction of a protein is sufficient for an NMR hit, whereas in X-ray crystallography it requires 30–70% binding to be considered as a potential hit [20][21][22]. Studies have also proposed that the quality of binding hits obtained from crystallography is more rigid and qualitative compared to techniques such as SPR, mass spectrometry, calorimetry, and NMR [23].
2.3. Modeling
During modeling procedures, smaller fragments, compared to the target enzyme, make molecular docking more difficult due to a lack of interactions with the residual molecules and low-affinity binding to the functional groups [8]. Under these conditions, the fragment analysis might turn out to be difficult as some potent fragment hits may have been missed due to weaker interactions and the smaller size of the protein. Thus, the smaller size of the fragments might not be promising candidates for docking unless they share related physical-chemical properties and binding pockets that eventually lead to the time-consuming subsequent analysis [24]. Among the modeling procedures chosen for FBDD, docking has advanced in recent years. Researchers have various options that can be chosen either from stoichiometric methods by following empirical or conformational changes, force-field, or using a systematic approach [25]. However, even though all of these factors were considered, the accuracy remains unclear due to the receptor–ligand binding efficacy. To avoid these conditions, standard protein models and potent binding sites are essential for FBDD. The majority of the membrane proteins, such as G protein-coupled receptors (GPCRs), do not have any information on crystallization, which is challenging in the development of new drugs [26]. Homology modeling has answered its insights into this problem by targeting proteins with homology sequences and crystallizing the targeted proteins using static models [27][28]. Homology modeling is dependent on changes in binding site conformation and the shape of the protein structure. Despite these drawbacks, artificial intelligence algorithms and new chemical informatics were also improved for in silico drug design using molecular dynamic simulations [21][29]. With this approach, potent drug candidates can be assessed early during fragment-based drug discovery.
2.4. Challenges in Hit Identification and Lead Optimization
Proteins encoded by essential genes play a crucial role in the survival of organisms. Several studies after the publication of the first Mtb genome sequence have determined essential genes and, using a target-based approach, they have designed novel inhibitors against them [7]. Understanding the molecular targets and modes of action is necessary to develop a drug for the treatment of tuberculosis without failure. In recent times, Mtb therapeutics has been focusing on identifying effective compounds that specifically inhibit the survival of the bacteria and proliferation in the host. The genome of Mtb encodes several virulent proteins that are secreted to alter the host’s immune system, which helps to improve resistance against therapeutics [30][31]. Therefore, it limits the pathogen’s activities in the host by inhibiting the essential pathogenic proteins. The key feature of the fragment-based approach is to isolate the protein in a larger quantity and maintain its stability for the screenings. Further, crystallization of the protein target is required to determine the binding modes. The fragment-based approach is difficult without stability and high yields of protein. Additionally, genetic screen trials are the first steps in identifying the genetic products that will be the focus of chemotherapy against Mtb. However, not all important genes are similarly responsive to pharmacological treatment [32]. The lower molecular weight compounds with inhibitor action must have specificity to affect the target function without the interference of any host orthologs [33][34].
The fragment-based approach is now an established and effective method to develop compounds that can inhibit or activate the function of a target protein. The small molecules (<18 heavy atoms) exhibit their functionality to bind and are small enough to fit into the active site of the protein, which is a major advantage over larger compounds that block the binding site of the protein molecule [35]. Fragment screening in the library is the first step to screening small molecules using various biophysical and functional assays. Another important screening approach is soaking the fragments with a known crystal structure of the protein, which is useful to detect weak binding and characterize the binding position of the fragment [36]. However, it has been challenging to prepare larger quantities of soaked crystals, collect the data in higher amounts, and complete the processing necessary to investigate the thousands of diffraction datasets to screen every single compound in the fragment library.
The screening of library fragments against an enzyme generates a significant number of hits. The absolute hit rate depends on the target, but usually most fragment screens result from multiple hits are more than 10, which can be developed into a different chemical series. The fragment growth strategy, used for developing the fragment hits, is effective when targeting the enclosed active site pockets and the effect of each functional group or atom is to be assessed thoroughly; hence, maintaining optimal ligand efficiency. It is preferable that the target protein should have compact sites that are capable of ligand-efficient interactions. In contrast, fragment linking might be responsible for a less ligand-efficient site that has multiple subpockets. For example, the potency fragments can be developed into potent inhibitors. For example, pantothenate synthase of Mtb, which is the logically direct way [37]. During each synthetic cycle, the analysis of enzyme–ligand complexes and ligand efficiencies is useful to guide the rational design. Therefore, there is no chance of incorrect assumptions during the optimization process. Similarly, the detailed structural analysis helps to guide the selection of linkers and in the understanding of the imposed conformational restraints. Although it appears more elegant, the repertoire of linkers is limited and compromises the binding of the original. On the other hand, flexibility will be present at each stage in the fragment-growing approach and will allow more space for further optimization.
2.5. Post-Modeling Expression, Solubilization, Purification, Crystallization, Data Collection, and Structure Solution
X-ray crystallography is an applicable technique to validate any hits during the screening process. The crystal structure reveals the binding mode of the fragment. However, crystallography requires bulk expression and a high amount of pure protein in a soluble form, which is challenging and time-consuming. Specifically, membrane proteins are difficult to crystallize due to the presence of hydrophobic transmembrane regions (TMRs). Crystal soaking is preferable as it leads to the high-affinity interactions. However, it needs further investigation. When the fragment is not soluble, then co-crystallization can be used, and only one fragment is crystallized with the target protein [38]. Crystal formation, data collection, and analysis are limiting factors for the FBDD approach, but robotic automation has made the overall crystallization process simpler and more convenient as it has made it feasible to work with smaller quantities of proteins.
The new generation of detectors is the fastest data collection tool. For example, they have termed pixel array detectors (PADs) an emerging technology and synchrotron beamlines widespread. Structures can be solved by molecular replacement using PHASER [39] and refinements can be achieved using REFMAC [40] and PHENIX [41]. Many programs are available to build ligands into electron densities. Commercial packages include PrimeX by Schrödinger (New York, NY, U.S.A.), Rhofit by Global Phasing (Cambridge, U.K.), and Afitt by OpenEye (Santa Fe, NM, U.S.A.) [42]. However, the major pitfalls of X-ray crystal structures are fragment screening and lead design, which depend on the skill of the crystallographer regardless of resolution. Uncertainty in individual atom positions and amino acid placements, changes in solvent structures, and solvent binding interpretation become more difficult.
References
- World Health Organization. Global Tuberculosis Report 2020; World Health Organization: Geneva, Switzerland, 2020; ISBN 978-92-4-001313-1.
- Tiberi, S.; du Plessis, N.; Walzl, G.; Vjecha, M.J.; Rao, M.; Ntoumi, F.; Mfinanga, S.; Kapata, N.; Mwaba, P.; McHugh, T.D.; et al. Tuberculosis: Progress and advances in development of new drugs, treatment regimens, and host-directed therapies. Lancet Infect. Dis. 2018, 18, e183–e198.
- Abell, C.; Dagostin, C. Chapter 1. Different Flavours of Fragments. In Fragment-Based Drug Discovery; Howard, S., Abell, C., Eds.; The Royal Society of Chemistry: London, UK, 2015; pp. 1–18.
- Jacquemard, C.; Kellenberger, E. A bright future for fragment-based drug discovery: What does it hold? Expert Opin. Drug Discov. 2019, 14, 413–416.
- Shuker, S.B.; Hajduk, P.J.; Meadows, R.P.; Fesik, S.W. Discovering High-Affinity Ligands for Proteins: SAR by NMR. Science 1996, 274, 1531–1534.
- Blundell, T.L.; Jhoti, H.; Abell, C. High-throughput crystallography for lead discovery in drug design. Nat. Rev. Drug Discov. 2002, 1, 45–54.
- Oh, S.; Trifonov, L.; Yadav, V.D.; Barry, C.E.; Boshoff, H.I. Tuberculosis Drug Discovery: A Decade of Hit Assessment for Defined Targets. Front. Cell. Infect. Microbiol. 2021, 11, 611304.
- Li, Q. Application of Fragment-Based Drug Discovery to Versatile Targets. Front. Mol. Biosci. 2020, 7, 180.
- Erlanson, D.A.; Fesik, S.W.; Hubbard, R.E.; Jahnke, W.; Jhoti, H. Twenty years on: The impact of fragments on drug discovery. Nat. Rev. Drug Discov. 2016, 15, 605–619.
- Ayotte, Y.; Bernet, E.; Bilodeau, F.; Cimino, M.; Gagnon, D.; Lebughe, M.; Mistretta, M.; Ogadinma, P.; Ouali, S.L.; Sow, A.A.; et al. Fragment-Based Phenotypic Lead Discovery to Identify New Drug Seeds That Target Infectious Diseases. ACS Chem. Biol. 2021, 16, 2158–2163.
- Charoensutthivarakul, S.; Thomas, S.E.; Curran, A.; Brown, K.P.; Belardinelli, J.M.; Whitehouse, A.J.; Acebrón-García-de-Eulate, M.; Sangan, J.; Gramani, S.G.; Jackson, M.; et al. Development of Inhibitors of SAICAR Synthetase (PurC) from Mycobacterium abscessus Using a Fragment-Based Approach. ACS Infect. Dis. 2022, 8, 296–309.
- Murray, C.W.; Verdonk, M.L.; Rees, D.C. Experiences in fragment-based drug discovery. Trends Pharmacol. Sci. 2012, 33, 224–232.
- Kirsch, P.; Hartman, A.M.; Hirsch, A.K.H.; Empting, M. Concepts and Core Principles of Fragment-Based Drug Design. Molecules 2019, 24, 4309.
- Jhoti, H.; Williams, G.; Rees, D.C.; Murray, C.W. The “rule of three” for fragment-based drug discovery: Where are we now? Nat. Rev. Drug Discov. 2013, 12, 644.
- Cramer, J.; Schiebel, J.; Wulsdorf, T.; Grohe, K.; Najbauer, E.E.; Ehrmann, F.R.; Radeva, N.; Zitzer, N.; Linne, U.; Linser, R.; et al. A False-Positive Screening Hit in Fragment-Based Lead Discovery: Watch out for the Red Herring. Angew. Chem. Int. Ed. 2017, 56, 1908–1913.
- Murray, C.W.; Rees, D.C. The rise of fragment-based drug discovery. Nat. Chem. 2009, 1, 187–192.
- Joseph-McCarthy, D. Challenges of fragment screening. J. Comput.-Aided Mol. Des. 2009, 23, 449–451.
- Vangrevelinghe, E.; Rudisser, S. Computational Approaches for Fragment Optimization. Curr. Comput.-Aided Drug Des. 2007, 3, 69–83.
- Mureddu, L.G.; Vuister, G.W. Fragment-Based Drug Discovery by NMR. Where Are the Successes and Where Can It Be Improved? Front. Mol. Biosci. 2022, 9, 834453.
- Bian, Y. The Research and Development of an Artificial Intelligence Integrated Fragment-Based Drug Design Platform for Small Molecule Drug Discovery. Ph.D. Thesis, University of Pittsburgh, Pittsburgh, PA, USA, 2021.
- Lundquist, K.P.; Panchal, V.; Gotfredsen, C.H.; Brenk, R.; Clausen, M.H. Fragment-Based Drug Discovery for RNA Targets. ChemMedChem 2021, 16, 2588–2603.
- Bhoj, P.S.; Bahekar, S.; Khatri, V.; Singh, N.; Togre, N.S.; Goswami, K.; Chandak, H.S.; Dash, D. Role of Glutathione in Chalcone Derivative Induced Apoptosis of Brugia malayi and its Possible Therapeutic Implication. Acta Parasitol. 2021, 66, 406–415.
- Erlanson, D.A.; Wells, J.A.; Braisted, A.C. Tethering: Fragment-Based Drug Discovery. Annu. Rev. Biophys. Biomol. Struct. 2004, 33, 199–223.
- Erlanson, D.A.; McDowell, R.S.; O’Brien, T. Fragment-Based Drug Discovery. J. Med. Chem. 2004, 47, 3463–3482.
- Konteatis, Z.D. In silico fragment-based drug design. Expert Opin. Drug Discov. 2010, 5, 1047–1065.
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schiöth, H.B.; Gloriam, D.E. Trends in GPCR drug discovery: New agents, targets and indications. Nat. Rev. Drug Discov. 2017, 16, 829–842.
- Muhammed, M.T.; Aki-Yalcin, E. Homology modeling in drug discovery: Overview, current applications, and future perspectives. Chem. Biol. Drug Des. 2019, 93, 12–20.
- Cavasotto, C.N.; Phatak, S.S. Homology modeling in drug discovery: Current trends and applications. Drug Discov. Today 2009, 14, 676–683.
- de Souza Neto, L.R.; Moreira-Filho, J.T.; Neves, B.J.; Maidana, R.L.B.R.; Guimarães, A.C.R.; Furnham, N.; Andrade, C.H.; Silva, F.P. In Silico Strategies to Support Fragment-to-Lead Optimization in Drug Discovery. Front. Chem. 2020, 8, 93.
- Chiliza, T.E.; Pillay, M.; Pillay, B. Identification of unique essential proteins from a M. tuberculosis F15/LAM4/KZN phage secretome library. Pathog. Dis. 2017, 75, ftx001.
- Lamichhane, G. Novel targets in M. tuberculosis: Search for new drugs. Trends Mol. Med. 2011, 17, 25–33.
- Cragg, G.M.; Newman, D.J. Natural products: A continuing source of novel drug leads. Biochim. Biophys. Acta-Gen. Subj. 2013, 1830, 3670–3695.
- Sarathy, J.P.; Zuccotto, F.; Hsinpin, H.; Sandberg, L.; Via, L.E.; Marriner, G.A.; Masquelin, T.; Wyatt, P.; Ray, P.; Dartois, V. Prediction of Drug Penetration in Tuberculosis Lesions. ACS Infect. Dis. 2016, 2, 552–563.
- Zheng, X.; Av-Gay, Y. New Era of TB Drug Discovery and Its Impact on Disease Management. Curr. Treat. Options Infect. Dis. 2016, 8, 299–310.
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Molecular Biology of the Cell; Garland Science: New York, NY, USA, 2002; ISBN 0-8153-3218-1.
- Robson-Tull, J. Biophysical screening in fragment-based drug design: A brief overview. Biosci. Horizons Int. J. Stud. Res. 2018, 11, hzy015.
- Hung, A.W.; Silvestre, H.L.; Wen, S.; Ciulli, A.; Blundell, T.L.; Abell, C. Application of Fragment Growing and Fragment Linking to the Discovery of Inhibitors of Mycobacterium tuberculosis Pantothenate Synthetase. Angew. Chem. Int. Ed. 2009, 48, 8452–8456.
- Hassell, A.M.; An, G.; Bledsoe, R.K.; Bynum, J.M.; Carter, H.L.; Deng, S.-J.J.; Gampe, R.T.; Grisard, T.E.; Madauss, K.P.; Nolte, R.T.; et al. Crystallization of protein–ligand complexes. Acta Crystallogr. Sect. D Biol. Crystallogr. 2007, 63, 72–79.
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674.
- Murshudov, G.N.; Skubák, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC 5 for the refinement of macromolecular crystal structures. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 355–367.
- Adams, P.D.; Afonine, P.V.; Bunkóczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.-W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 213–221.
- Chilingaryan, Z.; Yin, Z.; Oakley, A.J. Fragment-Based Screening by Protein Crystallography: Successes and Pitfalls. Int. J. Mol. Sci. 2012, 13, 12857–12879.
More
Information
Subjects:
Infectious Diseases
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.7K
Entry Collection:
Biopharmaceuticals Technology
Revisions:
2 times
(View History)
Update Date:
23 Sep 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No