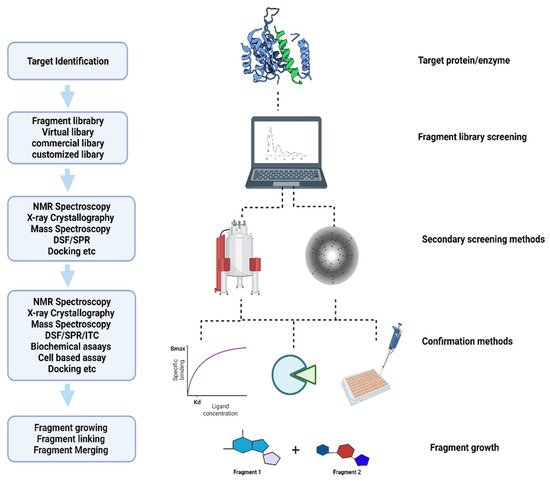
The World Health Organization (WHO) has reported global tuberculosis (TB) with an estimated 10 million active new cases, 1.5 million deaths, and 1.7 billion latent infections in 2020 [
1]. Although the effective treatment of TB has been available for over four decades, the occurrence of drug resistance (DR) and multi-drug resistance (MDR) is a major challenge during the treatment course. According to the recent WHO report, although the total number of DR cases decreased by 22% in 2019–2020, the existing drug regimens are ineffective in combating the disease progression [
1,
2]. Recent studies identified fragment-based drug discovery (FBDD) as a key approach in the field of drug discovery to find new targets through high-throughput screening (HTS) techniques [
3,
4]. The identification of even smaller molecules, the druggability of biological targets, and alternate inhibition sites on established drugs are key to the FBDD’s success [
5,
6]. Based on this approach, more than 40 new compounds have been identified and are currently in clinical trials [
7,
8,
9].
Figure 1 shows the basic approach used to identify new compounds in FBDD.
This FBDD approach has evolved steadily over the last decade. The application of fragment library screening has been successfully applied to a wide range of infectious diseases, including, but not limited to,
Leishmania, flaviviruses, non-tuberculosis mycobacteria (NTM), and Mtb [
10]. The discovery of the phosphoribosylaminoimidazole succinocarboxamide synthetase enzyme (PurC) as a potential inhibitor target in both
M. abscessus and Mtb is also a significant breakthrough in FBDD [
11]. It is crucial to understand how beneficial the FBDD approach is in developing new molecules with possible potential therapeutic activity against NTM and Mtb infections.
2. Challenges in Fragment-Based Drug Discovery
Drug discovery for NTMs and MTB has been successfully accomplished using the FBDD. This strategy, however, faces a number of obstacles and challenges with its methodologies. We have covered a lot of them in this area, which will help to overcome the challenges listed below and pave the way for successful drug discovery (Table 1).
Table 1. Challenges in the FBDD approach.
| Method/Molecules |
Challenges |
| X-ray crystallography |
- −
-
Bulk expression and solubility
- −
-
Crystal formation, data collection and analysis
|
| X-ray crystal structures |
- −
-
Fragment screening and lead design
- −
-
Positions of the atom, amino acid placements, changes in solvent structures, and solvent binding interpretations
|
| Crystal soaking |
- −
-
Further investigation for high-affinity interactions
- −
-
Prepare larger quantities of soaked crystals
|
| Molecular docking with smaller fragments |
- −
-
Low affinity binding
- −
-
Lack of interactions
- −
-
Fragment analysis
|
| Homology modeling |
- −
-
Conformational changes in binding sites and shape of the protein structure
|
| Stoichiometric approach |
- −
-
Accuracy of receptor–ligand binding efficiency
|
| Fragment screening |
- −
-
Large size or target and multiprotein complexes
|
| Fragments |
- −
-
Smaller size and weak affinity
- −
-
Flat structured fragments
|
| Low efficiency fragments |
- −
-
Optimization
|
| Fragment potential hits |
- −
-
Quality of binding
|
2.1. Specialized Methods Are Needed to Detect Fragment Binding in Libraries
The fragments form high-attribute interactions if they have suitable ligand efficiencies (LE), which are ineffective due to their smaller size and weak affinity [
138]. It is well known that selected fragment libraries follow the “rule of three”. For example, using three-dimensional fragments instead of flat structures, identifying differences at fragment binding sites and protein binding sites, and determining the potent target hits for fragment protein interaction. Although there are arguments against using the rule of three, it is considered the preferred model for fragment selection in libraries [
16,
139,
140]. Various approaches, such as NMR, surface plasmon resonance (SPR), and X-ray screening, have greater sensitivity and are specifically used to screen larger fragments in higher libraries in fragment-based drug discovery (FBDD) [
30]. Additionally, thermal denaturation, thermal electrophoresis, capillary electrophoresis, mass spectrometry, and isothermal titration calorimetry have also been widely used in FBDD despite their limitations.
2.2. Optimization of Fragment Hits Using Computational Tools
Computational tools, such as de novo design, combinatorial docking, and interactive optimization, and their approaches, play a key role in the optimization process through fragment linking that eventually reduces the time required for increasing the efficacy of potent fragment leads [
141,
142]. Optimization and proper knowledge of specific fragments is a pre-requisite for FBDD [
143]. The large size of targeted molecules or the existence of targets as multiprotein complexes significantly impacts the generation of robust crystals, leading to the hindrance of using the FBDD approach for screening fragments [
8]. The optimization of fragment hits also depends on detection techniques considered for FBDD; the correlation of fragment hits must be carefully considered. For example, 5% of fragment interaction of a protein is sufficient for an NMR hit, whereas in X-ray crystallography it requires 30–70% binding to be considered as a potential hit [
144,
145,
146]. Studies have also proposed that the quality of binding hits obtained from crystallography is more rigid and qualitative compared to techniques such as SPR, mass spectrometry, calorimetry, and NMR [
147].
2.3. Modeling
During modeling procedures, smaller fragments, compared to the target enzyme, make molecular docking more difficult due to a lack of interactions with the residual molecules and low-affinity binding to the functional groups [
8]. Under these conditions, the fragment analysis might turn out to be difficult as some potent fragment hits may have been missed due to weaker interactions and the smaller size of the protein. Thus, the smaller size of the fragments might not be promising candidates for docking unless they share related physical-chemical properties and binding pockets that eventually lead to the time-consuming subsequent analysis [
148]. Among the modeling procedures chosen for FBDD, docking has advanced in recent years. Researchers have various options that can be chosen either from stoichiometric methods by following empirical or conformational changes, force-field, or using a systematic approach [
149]. However, even though all of these factors were considered, the accuracy remains unclear due to the receptor–ligand binding efficacy. To avoid these conditions, standard protein models and potent binding sites are essential for FBDD. The majority of the membrane proteins, such as G protein-coupled receptors (GPCRs), do not have any information on crystallization, which is challenging in the development of new drugs [
150]. Homology modeling has answered its insights into this problem by targeting proteins with homology sequences and crystallizing the targeted proteins using static models [
151,
152]. Homology modeling is dependent on changes in binding site conformation and the shape of the protein structure. Despite these drawbacks, artificial intelligence algorithms and new chemical informatics were also improved for in silico drug design using molecular dynamic simulations [
145,
153]. With this approach, potent drug candidates can be assessed early during fragment-based drug discovery.
2.4. Challenges in Hit Identification and Lead Optimization
Proteins encoded by essential genes play a crucial role in the survival of organisms. Several studies after the publication of the first Mtb genome sequence have determined essential genes and, using a target-based approach, they have designed novel inhibitors against them [
7]. Understanding the molecular targets and modes of action is necessary to develop a drug for the treatment of tuberculosis without failure. In recent times, Mtb therapeutics has been focusing on identifying effective compounds that specifically inhibit the survival of the bacteria and proliferation in the host. The genome of Mtb encodes several virulent proteins that are secreted to alter the host’s immune system, which helps to improve resistance against therapeutics [
154,
155]. Therefore, it limits the pathogen’s activities in the host by inhibiting the essential pathogenic proteins. The key feature of the fragment-based approach is to isolate the protein in a larger quantity and maintain its stability for the screenings. Further, crystallization of the protein target is required to determine the binding modes. The fragment-based approach is difficult without stability and high yields of protein. Additionally, genetic screen trials are the first steps in identifying the genetic products that will be the focus of chemotherapy against Mtb. However, not all important genes are similarly responsive to pharmacological treatment [
156]. The lower molecular weight compounds with inhibitor action must have specificity to affect the target function without the interference of any host orthologs [
157,
158].
The fragment-based approach is now an established and effective method to develop compounds that can inhibit or activate the function of a target protein. The small molecules (<18 heavy atoms) exhibit their functionality to bind and are small enough to fit into the active site of the protein, which is a major advantage over larger compounds that block the binding site of the protein molecule [
159]. Fragment screening in the library is the first step to screening small molecules using various biophysical and functional assays. Another important screening approach is soaking the fragments with a known crystal structure of the protein, which is useful to detect weak binding and characterize the binding position of the fragment [
35]. However, it has been challenging to prepare larger quantities of soaked crystals, collect the data in higher amounts, and complete the processing necessary to investigate the thousands of diffraction datasets to screen every single compound in the fragment library.
The screening of library fragments against an enzyme generates a significant number of hits. The absolute hit rate depends on the target, but usually most fragment screens result from multiple hits are more than 10, which can be developed into a different chemical series. The fragment growth strategy, used for developing the fragment hits, is effective when targeting the enclosed active site pockets and the effect of each functional group or atom is to be assessed thoroughly; hence, maintaining optimal ligand efficiency. It is preferable that the target protein should have compact sites that are capable of ligand-efficient interactions. In contrast, fragment linking might be responsible for a less ligand-efficient site that has multiple subpockets. For example, the potency fragments can be developed into potent inhibitors. For example, pantothenate synthase of Mtb, which is the logically direct way [
66]. During each synthetic cycle, the analysis of enzyme–ligand complexes and ligand efficiencies is useful to guide the rational design. Therefore, there is no chance of incorrect assumptions during the optimization process. Similarly, the detailed structural analysis helps to guide the selection of linkers and in the understanding of the imposed conformational restraints. Although it appears more elegant, the repertoire of linkers is limited and compromises the binding of the original. On the other hand, flexibility will be present at each stage in the fragment-growing approach and will allow more space for further optimization.
2.5. Post-Modeling Expression, Solubilization, Purification, Crystallization, Data Collection, and Structure Solution
X-ray crystallography is an applicable technique to validate any hits during the screening process. The crystal structure reveals the binding mode of the fragment. However, crystallography requires bulk expression and a high amount of pure protein in a soluble form, which is challenging and time-consuming. Specifically, membrane proteins are difficult to crystallize due to the presence of hydrophobic transmembrane regions (TMRs). Crystal soaking is preferable as it leads to the high-affinity interactions. However, it needs further investigation. When the fragment is not soluble, then co-crystallization can be used, and only one fragment is crystallized with the target protein [
160]. Crystal formation, data collection, and analysis are limiting factors for the FBDD approach, but robotic automation has made the overall crystallization process simpler and more convenient as it has made it feasible to work with smaller quantities of proteins.
The new generation of detectors is the fastest data collection tool. For example, they have termed pixel array detectors (PADs) an emerging technology and synchrotron beamlines widespread. Structures can be solved by molecular replacement using PHASER [
161] and refinements can be achieved using REFMAC [
162] and PHENIX [
163]. Many programs are available to build ligands into electron densities. Commercial packages include PrimeX by Schrödinger (New York, NY, U.S.A.), Rhofit by Global Phasing (Cambridge, U.K.), and Afitt by OpenEye (Santa Fe, NM, U.S.A.) [
164]. However, the major pitfalls of X-ray crystal structures are fragment screening and lead design, which depend on the skill of the crystallographer regardless of resolution. Uncertainty in individual atom positions and amino acid placements, changes in solvent structures, and solvent binding interpretation become more difficult.