 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Gabriele Carullo | -- | 6235 | 2022-09-20 10:59:55 | | | |
| 2 | Beatrix Zheng | + 3 word(s) | 6238 | 2022-09-21 04:04:03 | | | | |
| 3 | Beatrix Zheng | Meta information modification | 6238 | 2022-09-23 04:45:08 | | |
Video Upload Options
Histone deacetylases (HDACs), also known as lysine deacetylases (KDACs), belong to the class of zinc (Zn2+)-dependent or nicotinamide adenine dinucleotide (NAD+)-dependent proteolytic enzymes. HDACs participate in transcriptional repression and chromatin condensation mechanisms by removing the acetyl moiety from the acetylated ε-amino group of histone lysines and other non-histone proteins. These enzymes play a pivotal role in the modulation of several cellular pathways such as cell proliferation, apoptosis, neurogenesis and epigenetic regulations. In some cases, HDACs are involved in the occurrence and progression of numerous pathophysiological conditions as well as diseases such as neurological disorders, fibrosis, cancer, metabolic dysfunctions and parasitic infections.
1. HDAC8 Is a Class I HDAC Enzyme
-
HDAC8 is an X-linked protein which acts independently, e.g., without forming any co-complexes for the activity;
-
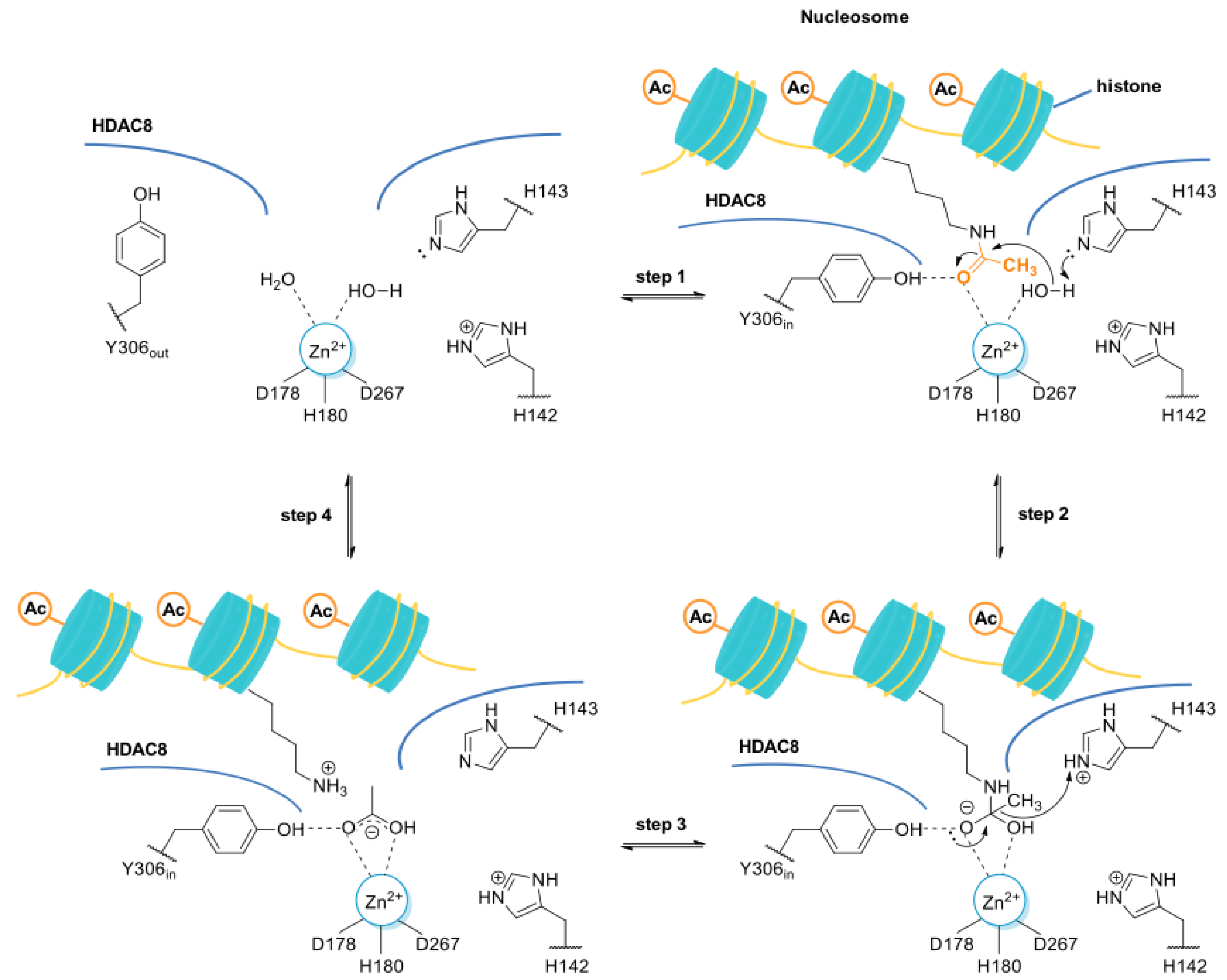
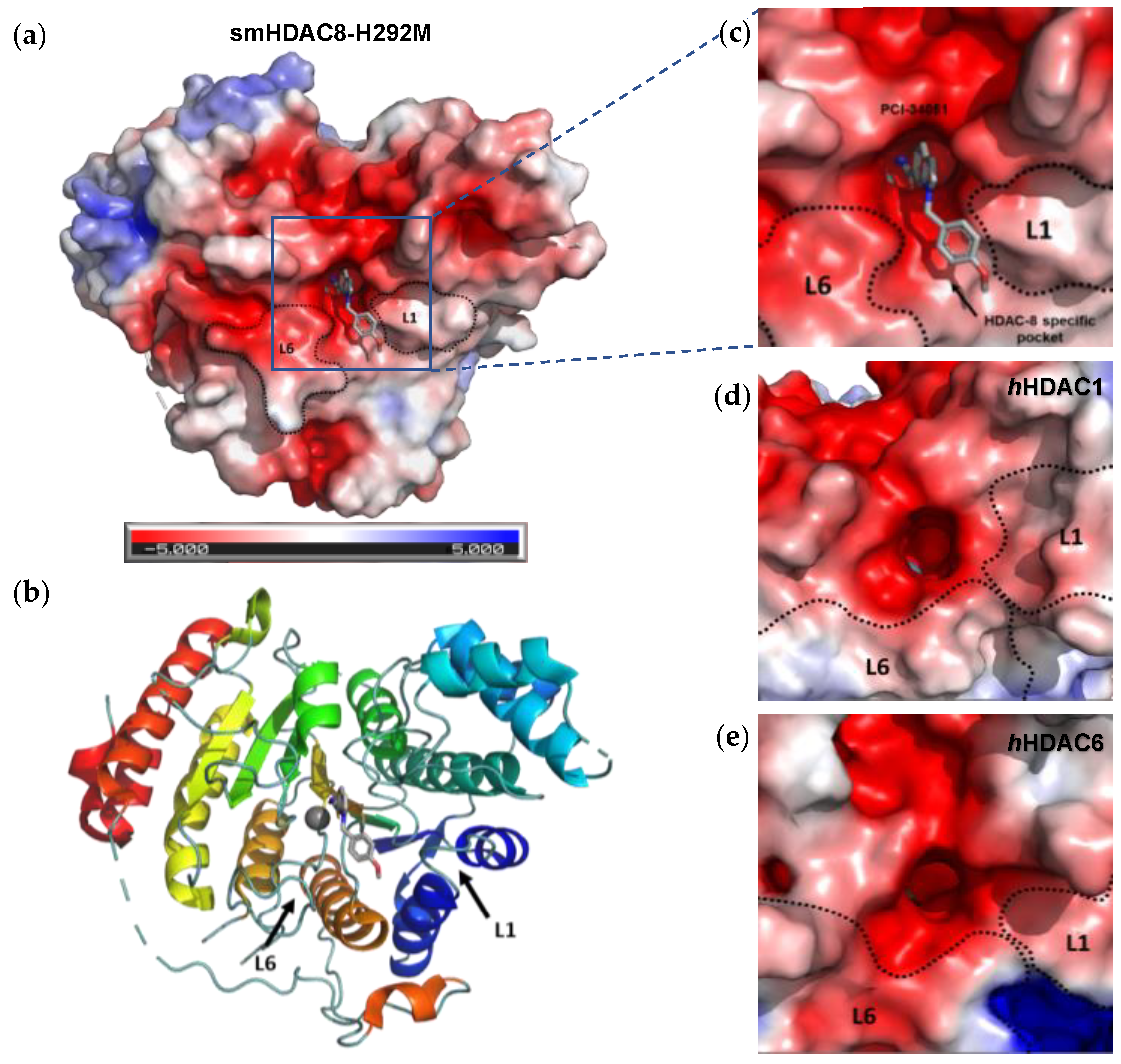
The L1 loop of HDAC8 is closest to the enzyme active site and undergoes conformational changes, differently depending on the substrate (Figure 2);
-
L1 and L6 form a specific pocket which requires an “L” shape conformation for selective binding (Figure 2);
-
HDAC8 presents a nuclear localization sequence between the catalytic domain of the enzyme and the serine binding motif found at the end of the catalytic domain [17].
1.1. HDAC8 Substrates
1.1.1. SMC3
1.1.2. p53
1.1.3. ERRα
1.1.4. inv(16)
1.1.5. CREB
2. Involvement of HDAC8 in Different Diseases
2.1. X-Linked Disorders
2.1.1. Cornelia de Lange Syndrome (CdLS)
2.1.2. Duchenne Muscular Dystrophy (DMD)
2.2. Aberrant Wound Healing
2.2.1. Pulmonary Fibrosis (PF)
2.2.2. Renal Fibrosis
2.2.3. Liver Fibrosis
2.2.4. Cardiac Fibrosis
2.2.5. Aberrant Wound Healing Associated with Diabetic Foot Ulcers (DFU)
2.3. Cancer
2.3.1. Haematological Malignancies
2.3.2. Solid Tumours
References
- Carullo, G.; Federico, S.; Relitti, N.; Gemma, S.; Butini, S.; Campiani, G. Retinitis Pigmentosa and Retinal Degenerations: Deciphering Pathways and Targets for Drug Discovery and Development. ACS Chem. Neurosci. 2020, 11, 2173–2191.
- Castañeda, C.A.; Wolfson, N.A.; Leng, K.R.; Kuo, Y.-M.; Andrews, A.J.; Fierke, C.A. HDAC8 substrate selectivity is determined by long- and short-range interactions leading to enhanced reactivity for full-length histone substrates compared with peptides. J. Biol. Chem. 2017, 292, 21568–21577.
- Chakrabarti, A.; Melesina, J.; Kolbinger, F.; Oehme, I.; Senger, J.; Witt, O.; Sippl, W.; Jung, M. Targeting histone deacetylase 8 as a therapeutic approach to cancer and neurodegenerative diseases. Futur. Med. Chem. 2016, 8, 1609–1634.
- Durst, K.L.; Lutterbach, B.; Kummalue, T.; Friedman, A.D.; Hiebert, S.W. The inv(16) Fusion Protein Associates with Corepressors via a Smooth Muscle Myosin Heavy-Chain Domain. Mol. Cell. Biol. 2003, 23, 607–619.
- Aramsangtienchai, P.; Spiegelman, N.; He, B.; Miller, S.P.; Dai, L.; Zhao, Y.; Lin, H. HDAC8 Catalyzes the Hydrolysis of Long Chain Fatty Acyl Lysine. ACS Chem. Biol. 2016, 11, 2685–2692.
- Porter, N.J.; Christianson, D.W. Structure, mechanism, and inhibition of the zinc-dependent histone deacetylases. Curr. Opin. Struct. Biol. 2019, 59, 9–18.
- Somoza, J.R.; Skene, R.; Katz, B.A.; Mol, C.; Ho, J.D.; Jennings, A.J.; Luong, C.; Arvai, A.; Buggy, J.J.; Chi, E.; et al. Structural Snapshots of Human HDAC8 Provide Insights into the Class I Histone Deacetylases. Structure 2004, 12, 1325–1334.
- Dowling, D.P.; Gantt, S.L.; Gattis, S.G.; Fierke, C.A.; Christianson, D.W. Structural Studies of Human Histone Deacetylase 8 and Its Site-Specific Variants Complexed with Substrate and Inhibitors. Biochemistry 2008, 47, 13554–13563.
- Vannini, A.; Volpari, C.; Gallinari, P.; Jones, P.; Mattu, M.; Carfí, A.; De Francesco, R.; Steinkühler, C.; Di Marco, S. Substrate binding to histone deacetylases as shown by the crystal structure of the HDAC8–substrate complex. EMBO Rep. 2007, 8, 879–884.
- Porter, N.J.; Christianson, N.H.; Decroos, C.; Christianson, D.W. Structural and Functional Influence of the Glycine-Rich Loop G302GGGY on the Catalytic Tyrosine of Histone Deacetylase 8. Biochemistry 2016, 55, 6718–6729.
- Marek, M.; Shaik, T.B.; Heimburg, T.; Chakrabarti, A.; Lancelot, J.; Morales, E.R.; Da Veiga, C.; Kalinin, D.; Melesina, J.; Robaa, D.; et al. Characterization of Histone Deacetylase 8 (HDAC8) Selective Inhibition Reveals Specific Active Site Structural and Functional Determinants. J. Med. Chem. 2018, 61, 10000–10016.
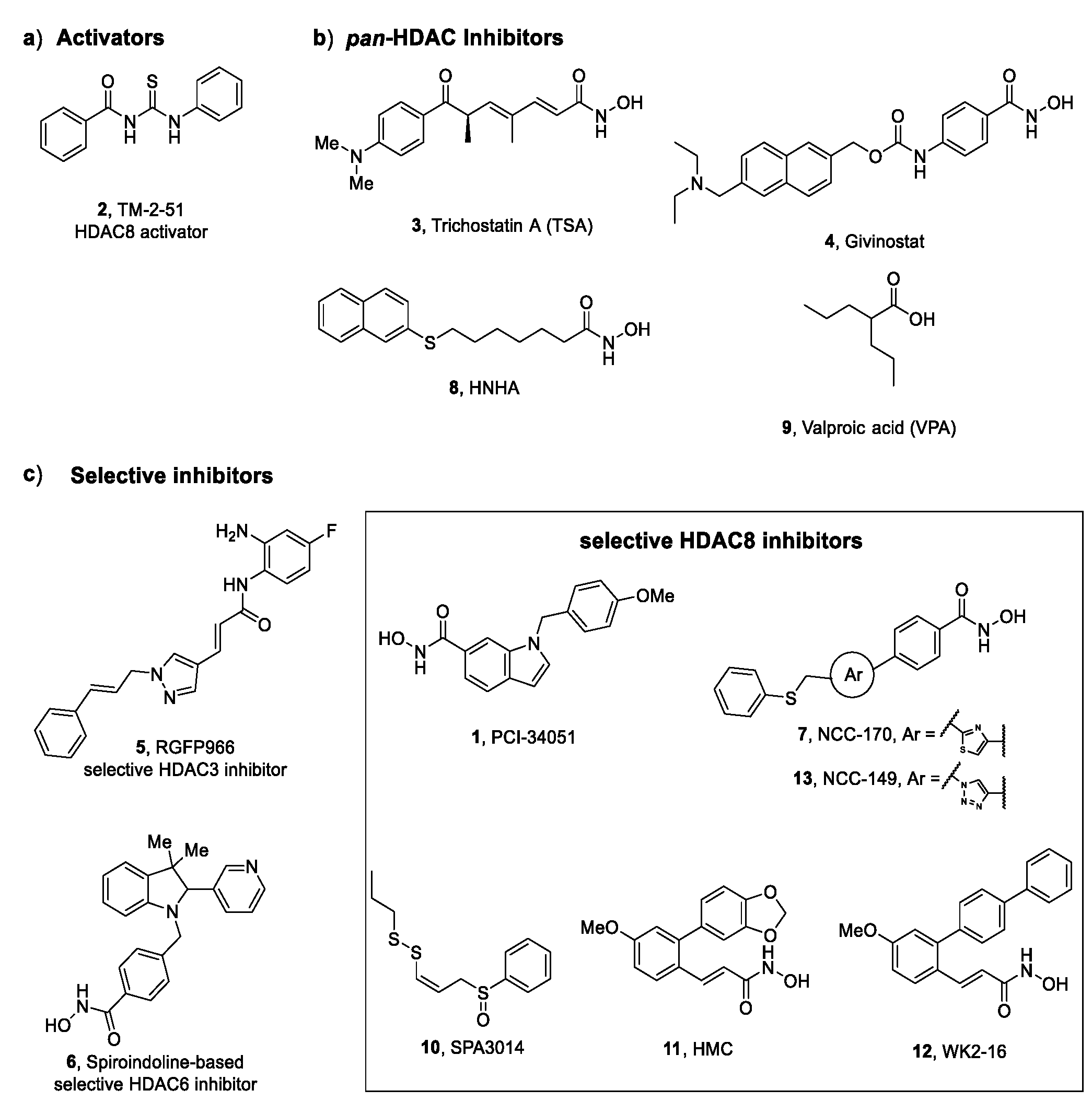
- Balasubramanian, S.; Ramos, J.; Luo, W.; Sirisawad, M.; Verner, E.; Buggy, J.J. A novel histone deacetylase 8 (HDAC8)-specific inhibitor PCI-34051 induces apoptosis in T-cell lymphomas. Leukemia 2008, 22, 1026–1034.
- Ho, T.C.S.; Chan, A.H.Y.; Ganesan, A. Thirty Years of HDAC Inhibitors: 2020 Insight and Hindsight. J. Med. Chem. 2020, 63, 12460–12484.
- Federico, S.; Khan, T.; Fontana, A.; Brogi, S.; Benedetti, R.; Sarno, F.; Carullo, G.; Pezzotta, A.; Saraswati, A.P.; Passaro, E.; et al. Azetidin-2-one-based small molecules as dual hHDAC6/HDAC8 inhibitors: Investigation of their mechanism of action and impact of dual inhibition profile on cell viability. Eur. J. Med. Chem. 2022, 238, 114409.
- Tabackman, A.A.; Frankson, R.; Marsan, E.S.; Perry, K.; Cole, K.E. Structure of ‘linkerless’ hydroxamic acid inhibitor-HDAC8 complex confirms the formation of an isoform-specific subpocket. J. Struct. Biol. 2016, 195, 373–378.
- Campiani, G.; Cavella, C.; Osko, J.D.; Brindisi, M.; Relitti, N.; Brogi, S.; Saraswati, A.P.; Federico, S.; Chemi, G.; Maramai, S.; et al. Harnessing the Role of HDAC6 in Idiopathic Pulmonary Fibrosis: Design, Synthesis, Structural Analysis, and Biological Evaluation of Potent Inhibitors. J. Med. Chem. 2021, 64, 9960–9988.
- Hassan, M.M.; Israelian, J.; Nawar, N.; Ganda, G.; Manaswiyoungkul, P.; Raouf, Y.S.; Armstrong, D.; Sedighi, A.; Olaoye, O.O.; Erdogan, F.; et al. Characterization of Conformationally Constrained Benzanilide Scaffolds for Potent and Selective HDAC8 Targeting. J. Med. Chem. 2020, 63, 8634–8648.
- Riester, D.; Hildmann, C.; Grünewald, S.; Beckers, T.; Schwienhorst, A. Factors affecting the substrate specificity of histone deacetylases. Biochem. Biophys. Res. Commun. 2007, 357, 439–445.
- Dose, A.; Liokatis, S.; Theillet, F.-X.; Selenko, P.; Schwarzer, D. NMR Profiling of Histone Deacetylase and Acetyl-transferase Activities in Real Time. ACS Chem. Biol. 2011, 6, 419–424.
- Chakrabarti, A.; Oehme, I.; Witt, O.; Oliveira, G.; Sippl, W.; Romier, C.; Pierce, R.J.; Jung, M. HDAC8: A multifaceted target for therapeutic interventions. Trends Pharmacol. Sci. 2015, 36, 481–492.
- Dasgupta, T.; Antony, J.; Braithwaite, A.W.; Horsfield, J.A. HDAC8 Inhibition Blocks SMC3 Deacetylation and Delays Cell Cycle Progression without Affecting Cohesin-dependent Transcription in MCF7 Cancer Cells. J. Biol. Chem. 2016, 291, 12761–12770.
- Yan, W.; Liu, S.; Xu, E.; Zhang, J.; Zhang, Y.; Chen, X. Histone deacetylase inhibitors suppress mutant p53 transcription via histone deacetylase 8. Oncogene 2013, 32, 599–609.
- Wilson, B.J.; Tremblay, A.M.; Deblois, G.; Sylvain-Drolet, G.; Giguere, V. An Acetylation Switch Modulates the Transcriptional Activity of Estrogen-Related Receptor α. Mol. Endocrinol. 2010, 24, 1349–1358.
- Gurard-Levin, Z.A.; Kim, J.; Mrksich, M. Combining Mass Spectrometry and Peptide Arrays to Profile the Specificities of Histone Deacetylases. ChemBioChem 2009, 10, 2159–2161.
- Karolczak-Bayatti, M.; Sweeney, M.; Cheng, J.; Edey, L.; Robson, S.C.; Ulrich, S.M.; Treumann, A.; Taggart, M.J.; Europe-Finner, G.N. Acetylation of Heat Shock Protein 20 (Hsp20) Regulates Human Myometrial Activity. J. Biol. Chem. 2011, 286, 34346–34355.
- Wolfson, N.A.; Pitcairn, C.A.; Fierke, C.A. HDAC8 substrates: Histones and beyond. Biopolymers 2013, 99, 112–126.
- Zhao, C.; Zang, J.; Ding, Q.; Inks, E.S.; Xu, W.; Chou, C.J.; Zhang, Y. Discovery of meta-sulfamoyl N-hydroxybenzamides as HDAC8 selective inhibitors. Eur. J. Med. Chem. 2018, 150, 282–291.
- Negmeldin, A.T.; Pflum, M.K.H. The structural requirements of histone deacetylase inhibitors: SAHA analogs modified at the C5 position display dual HDAC6/8 selectivity. Bioorganic Med. Chem. Lett. 2017, 27, 3254–3258.
- Tang, G.; Wong, J.C.; Zhang, W.; Wang, Z.; Zhang, N.; Peng, Z.; Zhang, Z.; Rong, Y.; Li, S.; Zhang, M.; et al. Identification of a Novel Aminotetralin Class of HDAC6 and HDAC8 Selective Inhibitors. J. Med. Chem. 2014, 57, 8026–8034.
- Chiu, C.-F.; Chin, H.-K.; Huang, W.-J.; Bai, L.-Y.; Huang, H.-Y.; Weng, J.-R. Induction of Apoptosis and Autophagy in Breast Cancer Cells by a Novel HDAC8 Inhibitor. Biomolecules 2019, 9, 824.
- Gao, X.; Huang, Z.; Fan, Y.; Sun, Y.; Liu, H.; Wang, L.; Gu, X.-F.; Yu, Y. A Functional Mutation in HDAC8 Gene as Novel Diagnostic Marker for Cornelia De Lange Syndrome. Cell. Physiol. Biochem. 2018, 47, 2388–2395.
- Du, J.; Li, W.; Liu, B.; Zhang, Y.; Yu, J.; Hou, X.; Fang, H. An in silico mechanistic insight into HDAC8 activation facilitates the discovery of new small-molecule activators. Bioorganic Med. Chem. 2020, 28, 115607.
- Elangkovan, N.; Dickson, G. Gene Therapy for Duchenne Muscular Dystrophy. J. Neuromuscul. Dis. 2021, 8, S303–S316.
- Sarogni, P.; Pallotta, M.M.; Musio, A. Cornelia de Lange syndrome: From molecular diagnosis to therapeutic approach. J. Med Genet. 2020, 57, 289–295.
- Ramos, F.J.; Puisac, B.; Baquero-Montoya, C.; Gil-Rodríguez, M.C.; Bueno, I.; Deardorff, M.A.; Hennekam, R.C.; Kaiser, F.J.; Krantz, I.D.; Musio, A.; et al. Clinical utility gene card for: Cornelia de Lange syndrome. Eur. J. Hum. Genet. 2015, 23, 1431.
- Deardorff, M.A.; Bando, M.; Nakato, R.; Watrin, E.; Itoh, T.; Minamino, M.; Saitoh, K.; Komata, M.; Katou, Y.; Clark, D.; et al. HDAC8 mutations in Cornelia de Lange syndrome affect the cohesin acetylation cycle. Nature 2012, 489, 313–317.
- Kaiser, F.J.; Ansari, M.; Braunholz, D.; Gil-Rodríguez, M.C.; Decroos, C.; Wilde, J.J.; Fincher, C.T.; Kaur, M.; Bando, M.; Amor, D.J.; et al. Loss-of-function HDAC8 mutations cause a phenotypic spectrum of Cornelia de Lange syndrome-like features, ocular hypertelorism, large fontanelle and X-linked inheritance. Hum. Mol. Genet. 2014, 23, 2888–2900.
- Kline, A.D.; Moss, J.; Selicorni, A.; Bisgaard, A.-M.; Deardorff, M.A.; Gillett, P.M.; Ishman, S.L.; Kerr, L.M.; Levin, A.V.; Mulder, P.A.; et al. Diagnosis and management of Cornelia de Lange syndrome: First international consensus statement. Nat. Rev. Genet. 2018, 19, 649–666.
- Singh, R.K.; Mandal, T.; Balsubramanian, N.; Viaene, T.; Leedahl, T.; Sule, N.; Cook, G.; Srivastava, D. Histone deacetylase activators: N-acetylthioureas serve as highly potent and isozyme selective activators for human histone deacetylase-8 on a fluorescent substrate. Bioorganic Med. Chem. Lett. 2011, 21, 5920–5923.
- Decroos, C.; Bowman, C.M.; Moser, J.-A.S.; Christianson, K.E.; Deardorff, M.A.; Christianson, D.W. Compromised Structure and Function of HDAC8 Mutants Identified in Cornelia de Lange Syndrome Spectrum Disorders. ACS Chem. Biol. 2014, 9, 2157–2164.
- Goemans, N.; Buyse, G. Current Treatment and Management of Dystrophinopathies. Curr. Treat. Options Neurol. 2014, 16, 287.
- Duan, D.; Goemans, N.; Takeda, S.; Mercuri, E.; Aartsma-Rus, A. Duchenne Muscular Dystrophy. Nat. Rev. Dis. Primers 2021, 7, 13.
- Minetti, G.C.; Colussi, C.; Adami, R.; Serra, C.; Mozzetta, C.; Parente, V.; Fortuni, S.; Straino, S.; Sampaolesi, M.; Di Padova, M.; et al. Functional and morphological recovery of dystrophic muscles in mice treated with deacetylase inhibitors. Nat. Med. 2006, 12, 1147–1150.
- Johnson, N.M.; Farr, G.H.; Maves, L. The HDAC Inhibitor TSA Ameliorates a Zebrafish Model of Duchenne Muscular Dystrophy. PLoS Curr. 2013, 5.
- Consalvi, S.; Mozzetta, C.; Bettica, P.; Germani, M.; Fiorentini, F.; Del Bene, F.; Rocchetti, M.; Leoni, F.; Monzani, V.; Mascagni, P.; et al. Preclinical Studies in the mdx Mouse Model of Duchenne Muscular Dystrophy with the Histone Deacetylase Inhibitor Givinostat. Mol. Med. 2013, 19, 79–87.
- Licandro, S.A.; Crippa, L.; Pomarico, R.; Perego, R.; Fossati, G.; Leoni, F.; Steinkühler, C. The pan HDAC inhibitor Givinostat improves muscle function and histological parameters in two Duchenne muscular dystrophy murine models expressing different haplotypes of the LTBP4 gene. Skelet. Muscle 2021, 11, 19.
- Spreafico, M.; Cafora, M.; Bragato, C.; Capitanio, D.; Marasca, F.; Bodega, B.; De Palma, C.; Mora, M.; Gelfi, C.; Marozzi, A.; et al. Targeting HDAC8 to ameliorate skeletal muscle differentiation in Duchenne muscular dystrophy. Pharmacol. Res. 2021, 170, 105750.
- Artlett, C.M. Inflammasomes in wound healing and fibrosis. J. Pathol. 2013, 229, 157–167.
- Pardali, E.; Sanchez-Duffhues, G.; Gomez-Puerto, M.C.; Dijke, P.T. TGF-β-Induced Endothelial-Mesenchymal Transition in Fibrotic Diseases. Int. J. Mol. Sci. 2017, 18, 2157.
- Kendall, R.T.; Feghali-Bostwick, C.A. Fibroblasts in fibrosis: Novel roles and mediators. Front. Pharmacol. 2014, 5, 123.
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 2012, 18, 1028–1040.
- Feng, Y.; Chen, D.; Vaziri, N.D.; Guo, Y.; Zhao, Y. Small molecule inhibitors of epithelial-mesenchymal transition for the treatment of cancer and fibrosis. Med. Res. Rev. 2020, 40, 54–78.
- Yoon, S.; Kang, G.; Eom, G.H. HDAC Inhibitors: Therapeutic Potential in Fibrosis-Associated Human Diseases. Int. J. Mol. Sci. 2019, 20, 1329.
- Miao, H.; Wu, X.-Q.; Zhang, D.-D.; Wang, Y.-N.; Guo, Y.; Li, P.; Xiong, Q.; Zhao, Y.-Y. Deciphering the cellular mechanisms underlying fibrosis-associated diseases and therapeutic avenues. Pharmacol. Res. 2021, 163, 105316.
- Pang, M.; Zhuang, S. Histone Deacetylase: A Potential Therapeutic Target for Fibrotic Disorders. J. Pharmacol. Exp. Ther. 2010, 335, 266–272.
- Hull, E.E.; Montgomery, M.R.; Leyva, K.J. HDAC Inhibitors as Epigenetic Regulators of the Immune System: Impacts on Cancer Therapy and Inflammatory Diseases. BioMed Res. Int. 2016, 2016, 8797206.
- Guo, W.; Shan, B.; Klingsberg, R.C.; Qin, X.; Lasky, J.A. Abrogation of TGF-β1-induced fibroblast-myofibroblast differentiation by histone deacetylase inhibition. Am. J. Physiol. Cell. Mol. Physiol. 2009, 297, L864–L870.
- Korfei, M.; Mahavadi, P.; Guenther, A. Targeting Histone Deacetylases in Idiopathic Pulmonary Fibrosis: A Future Therapeutic Option. Cells 2022, 11, 1626.
- Mathai, S.K.; Schwartz, D.A. Translational research in pulmonary fibrosis. Transl. Res. 2019, 209, 1–13.
- Chen, F.; Gao, Q.; Zhang, L.; Ding, Y.; Wang, H.; Cao, W. Inhibiting HDAC3 (Histone Deacetylase 3) Aberration and the Resultant Nrf2 (Nuclear Factor Erythroid-Derived 2-Related Factor-2) Repression Mitigates Pulmonary Fibrosis. Hypertension 2021, 78, e15–e25.
- Saito, S.; Zhuang, Y.; Suzuki, T.; Ota, Y.; Bateman, M.E.; Alkhatib, A.; Morris, G.F.; Lasky, J.A. HDAC8 inhibition ameliorates pulmonary fibrosis. Am. J. Physiol. Cell. Mol. Physiol. 2019, 316, L175–L186.
- François, H.; Chatziantoniou, C. Renal fibrosis: Recent translational aspects. Matrix Biol. 2018, 68–69, 318–332.
- Zhang, Y.; Meng, X.-M.; Huang, X.-R.; Lan, H.Y. The preventive and therapeutic implication for renal fibrosis by targetting TGF-β/Smad3 signaling. Clin. Sci. 2018, 132, 1403–1415.
- Yan, H.; Xu, J.; Xu, Z.; Yang, B.; Luo, P.; He, Q. Defining therapeutic targets for renal fibrosis: Exploiting the biology of pathogenesis. Biomed. Pharmacother. 2021, 143, 112115.
- Long, K.; Vaughn, Z.; McDaniels, M.D.; Joyasawal, S.; Przepiorski, A.; Parasky, E.; Sander, V.; Close, D.; Johnston, P.A.; Davidson, A.J.; et al. Validation of HDAC8 Inhibitors as Drug Discovery Starting Points to Treat Acute Kidney Injury. ACS Pharmacol. Transl. Sci. 2022, 5, 207–215.
- Zhang, Y.; Zou, J.; Tolbert, E.; Zhao, T.C.; Bayliss, G.; Zhuang, S. Identification of histone deacetylase 8 as a novel therapeutic target for renal fibrosis. FASEB J. 2020, 34, 7295–7310.
- Van Beneden, K.; Mannaerts, I.; Pauwels, M.; Branden, C.V.D.; van Grunsven, L.A. HDAC inhibitors in experimental liver and kidney fibrosis. Fibrogenesis Tissue Repair 2013, 6, 1.
- Park, K.C.; Park, J.H.; Jeon, J.Y.; Kim, S.Y.; Kim, J.M.; Lim, C.Y.; Lee, T.H.; Kim, H.K.; Lee, H.G.; Kwon, H.J.; et al. A new histone deacetylase inhibitor improves liver fibrosis inBDLrats through suppression of hepatic stellate cells. Br. J. Pharmacol. 2014, 171, 4820–4830.
- Aher, J.S.; Khan, S.; Jain, S.; Tikoo, K.; Jena, G. Valproate ameliorates thioacetamide-induced fibrosis by hepatic stellate cell inactivation. Hum. Exp. Toxicol. 2015, 34, 44–55.
- Lu, P.; Yan, M.; He, L.; Li, J.; Ji, Y.; Ji, J. Crosstalk between Epigenetic Modulations in Valproic Acid Deactivated Hepatic Stellate Cells: An Integrated Protein and miRNA Profiling Study. Int. J. Biol. Sci. 2019, 15, 93–104.
- Lee, C.H.; Choi, Y.; Cho, H.; Bang, I.H.; Hao, L.; Lee, S.-O.; Jeon, R.; Bae, E.J.; Park, B.-H. Histone deacetylase 8 inhibition alleviates cholestatic liver injury and fibrosis. Biochem. Pharmacol. 2021, 183, 114312.
- Lyu, X.; Hu, M.; Peng, J.; Zhang, X.; Sanders, Y.Y. HDAC inhibitors as antifibrotic drugs in cardiac and pulmonary fibrosis. Ther. Adv. Chronic Dis. 2019, 10, 2040622319862697.
- Liu, F.; Levin, M.D.; Petrenko, N.B.; Lu, M.M.; Wang, T.; Yuan, L.J.; Stout, A.L.; Epstein, J.A.; Patel, V.V. Histone-deacetylase inhibition reverses atrial arrhythmia inducibility and fibrosis in cardiac hypertrophy independent of angiotensin. J. Mol. Cell. Cardiol. 2008, 45, 715–723.
- Kang, S.-H.; Seok, Y.M.; Song, M.-J.; Lee, H.-A.; Kurz, T.; Kim, I. Histone Deacetylase Inhibition Attenuates Cardiac Hypertrophy and Fibrosis through Acetylation of Mineralocorticoid Receptor in Spontaneously Hypertensive Rats. Mol. Pharmacol. 2015, 87, 782–791.
- Zhao, T.; Kee, H.J.; Bai, L.; Kim, M.-K.; Kee, S.-J.; Jeong, M.H. Selective HDAC8 Inhibition Attenuates Isoproterenol-Induced Cardiac Hypertrophy and Fibrosis via p38 MAPK Pathway. Front. Pharmacol. 2021, 12, 677757.
- Zhao, T.; Kee, H.J.; Kee, S.-J.; Jeong, M.H. Hdac8 Inhibitor Alleviates Transverse Aortic Constriction-Induced Heart Failure in Mice by Downregulating Ace1. Oxidative Med. Cell. Longev. 2022, 2022, 6227330.
- Carullo, G.; Governa, P.; Leo, A.; Gallelli, L.; Citraro, R.; Cione, E.; Caroleo, M.C.; Biagi, M.; Aiello, F.; Manetti, F. Quercetin-3-Oleate Contributes to Skin Wound Healing Targeting FFA1/GPR40. ChemistrySelect 2019, 4, 8429–8433.
- Carullo, G.; Sciubba, F.; Governa, P.; Mazzotta, S.; Frattaruolo, L.; Grillo, G.; Cappello, A.R.; Cravotto, G.; Di Cocco, M.E.; Aiello, F. Mantonico and Pecorello Grape Seed Extracts: Chemical Characterization and Evaluation of In Vitro Wound-Healing and Anti-Inflammatory Activities. Pharmaceuticals 2020, 13, 97.
- Mazzotta, S.; Governa, P.; Borgonetti, V.; Marcolongo, P.; Nanni, C.; Gamberucci, A.; Manetti, F.; Pessina, F.; Carullo, G.; Brizzi, A.; et al. Pinocembrin and its linolenoyl ester derivative induce wound healing activity in HaCaT cell line potentially involving a GPR120/FFA4 mediated pathway. Bioorganic Chem. 2021, 108, 104657.
- Fu, W.; Liang, D.; Wu, X.; Chen, H.; Hong, X.; Wang, J.; Zhu, T.; Zeng, T.; Lin, W.; Chen, S.; et al. Long noncoding RNA LINC01435 impedes diabetic wound healing by facilitating YY1-mediated HDAC8 expression. iScience 2022, 25, 104006.
- Li, Y.; Sun, R.; Zou, J.; Ying, Y.; Luo, Z. Dual Roles of the AMP-Activated Protein Kinase Pathway in Angiogenesis. Cells 2019, 8, 752.
- Begum, F.; Keni, R.; Ahuja, T.N.; Beegum, F.; Nandakumar, K.; Shenoy, R.R. Notch signaling: A possible therapeutic target and its role in diabetic foot ulcers. Diabetes Metab. Syndr. Clin. Res. Rev. 2022, 16, 102542.
- Bhagat, T.D.; Zou, Y.; Huang, S.; Park, J.; Palmer, M.B.; Hu, C.; Li, W.; Shenoy, N.; Giricz, O.; Choudhary, G.; et al. Notch Pathway Is Activated via Genetic and Epigenetic Alterations and Is a Therapeutic Target in Clear Cell Renal Cancer. J. Biol. Chem. 2017, 292, 837–846.
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414.
- Banerjee, S.; Adhikari, N.; Amin, S.A.; Jha, T. Histone deacetylase 8 (HDAC8) and its inhibitors with selectivity to other isoforms: An overview. Eur. J. Med. Chem. 2019, 164, 214–240.
- Pantelaiou-Prokaki, G.; Mieczkowska, I.; Schmidt, G.E.; Fritzsche, S.; Prokakis, E.; Gallwas, J.; Wegwitz, F. HDAC8 suppresses the epithelial phenotype and promotes EMT in chemotherapy-treated basal-like breast cancer. Clin. Epigenetics 2022, 14, 7.
- Spreafico, M.; Gruszka, A.M.; Valli, D.; Mazzola, M.; Deflorian, G.; Quintè, A.; Totaro, M.G.; Battaglia, C.; Alcalay, M.; Marozzi, A.; et al. HDAC8: A Promising Therapeutic Target for Acute Myeloid Leukemia. Front. Cell Dev. Biol. 2020, 8, 844.
- Watters, J.M.; Wright, G.; Smith, M.A.; Shah, B.; Wright, K.L. Histone deacetylase 8 inhibition suppresses mantle cell lymphoma viability while preserving natural killer cell function. Biochem. Biophys. Res. Commun. 2021, 534, 773–779.
- An, P.; Chen, F.; Li, Z.; Ling, Y.; Peng, Y.; Zhang, H.; Li, J.; Chen, Z.; Wang, H. HDAC8 promotes the dissemination of breast cancer cells via AKT/GSK-3β/Snail signals. Oncogene 2020, 39, 4956–4969.
- Vanaja, G.R.; Ramulu, H.G.; Kalle, A.M. Overexpressed HDAC8 in cervical cancer cells shows functional redundancy of tubulin deacetylation with HDAC6. Cell Commun. Signal. 2018, 16, 20.
- Zheng, H.; Zhao, W.; Yan, C.; Watson, C.C.; Massengill, M.; Xie, M.; Massengill, C.; Noyes, D.R.; Martinez, G.V.; Afzal, R.; et al. HDAC Inhibitors Enhance T-Cell Chemokine Expression and Augment Response to PD-1 Immunotherapy in Lung Adenocarcinoma. Clin. Cancer Res. 2016, 22, 4119–4132.
- Beg, A.A.; Gray, J.E. HDAC inhibitors with PD-1 blockade: A promising strategy for treatment of multiple cancer types? Epigenomics 2016, 8, 1015–1017.
- Stone, M.L.; Chiappinelli, K.B.; Li, H.; Murphy, L.M.; Travers, M.E.; Topper, M.J.; Mathios, D.; Lim, M.; Shih, I.-M.; Wang, T.-L.; et al. Epigenetic therapy activates type I interferon signaling in murine ovarian cancer to reduce immunosuppression and tumor burden. Proc. Natl. Acad. Sci. USA 2017, 114, E10981–E10990.
- Briere, D.; Sudhakar, N.; Woods, D.M.; Hallin, J.; Engstrom, L.D.; Aranda, R.; Chiang, H.; Sodré, A.L.; Olson, P.; Weber, J.S.; et al. The class I/IV HDAC inhibitor mocetinostat increases tumor antigen presentation, decreases immune suppressive cell types and augments checkpoint inhibitor therapy. Cancer Immunol. Immunother. 2018, 67, 381–392.
- Knox, T.; Sahakian, E.; Banik, D.; Hadley, M.; Palmer, E.; Noonepalle, S.; Kim, J.; Powers, J.; Gracia-Hernandez, M.; Oliveira, V.; et al. Selective HDAC6 inhibitors improve anti-PD-1 immune checkpoint blockade therapy by decreasing the anti-inflammatory phenotype of macrophages and down-regulation of immunosuppressive proteins in tumor cells. Sci. Rep. 2019, 9, 6136.
- Maharaj, K.; Powers, J.J.; Mediavilla-Varela, M.; Achille, A.; Gamal, W.; Quayle, S.; Jones, S.S.; Sahakian, E.; Pinilla-Ibarz, J. HDAC6 Inhibition Alleviates CLL-Induced T-Cell Dysfunction and Enhances Immune Checkpoint Blockade Efficacy in the Eμ-TCL1 Model. Front. Immunol. 2020, 11, 590072.
- Burke, B.; Eden, C.; Perez, C.; Belshoff, A.; Hart, S.; Plaza-Rojas, L.; Delos Reyes, M.; Prajapati, K.; Voelkel-Johnson, C.; Henry, E.; et al. Inhibition of Histone Deacetylase (HDAC) Enhances Checkpoint Blockade Efficacy by Rendering Bladder Cancer Cells Visible for T Cell-Mediated Destruction. Front. Oncol. 2020, 10, 699.
- Baretti, M.; Yarchoan, M. Epigenetic modifiers synergize with immune-checkpoint blockade to enhance long-lasting antitumor efficacy. J. Clin. Investig. 2021, 131, e151002.
- Borcoman, E.; Kamal, M.; Marret, G.; Dupain, C.; Castel-Ajgal, Z.; Le Tourneau, C. HDAC Inhibition to Prime Immune Checkpoint Inhibitors. Cancers 2021, 14, 66.
- Yang, W.; Feng, Y.; Zhou, J.; Cheung, O.K.-W.; Cao, J.; Wang, J.; Tang, W.; Tu, Y.; Xu, L.; Wu, F.; et al. A selective HDAC8 inhibitor potentiates antitumor immunity and efficacy of immune checkpoint blockade in hepatocellular carcinoma. Sci. Transl. Med. 2021, 13, eaaz6804.
- Mormino, A.; Cocozza, G.; Fontemaggi, G.; Valente, S.; Esposito, V.; Santoro, A.; Bernardini, G.; Santoni, A.; Fazi, F.; Mai, A.; et al. Histone-deacetylase 8 drives the immune response and the growth of glioma. Glia 2021, 69, 2682–2698.
- Santos-Barriopedro, I.; Li, Y.; Bahl, S.; Seto, E. HDAC8 Affects MGMT Levels in Glioblastoma Cell Lines via Interaction with the Proteasome Receptor ADRM1. Genes Cancer 2019, 10, 119.
- Oehme, I.; Deubzer, H.E.; Wegener, D.; Pickert, D.; Linke, J.-P.; Hero, B.; Kopp-Schneider, A.; Westermann, F.; Ulrich, S.M.; von Deimling, A.; et al. Histone Deacetylase 8 in Neuroblastoma Tumorigenesis. Clin. Cancer Res. 2009, 15, 91–99.
- Rettig, I.; Koeneke, E.; Trippel, F.; Mueller, W.C.; Burhenne, J.; Kopp-Schneider, A.; Fabian, J.; Schober, A.; Fernekorn, U.; Von Deimling, A.; et al. Selective inhibition of HDAC8 decreases neuroblastoma growth in vitro and in vivo and enhances retinoic acid-mediated differentiation. Cell Death Dis. 2015, 6, e1657.
- Shen, J.; Najafi, S.; Stäble, S.; Fabian, J.; Koeneke, E.; Kolbinger, F.; Wrobel, J.K.; Meder, B.; Distel, M.; Heimburg, T.; et al. A kinome-wide RNAi screen identifies ALK as a target to sensitize neuroblastoma cells for HDAC8-inhibitor treatment. Cell Death Differ. 2018, 25, 2053–2070.

