1. HDAC8 Is a Class I HDAC Enzyme
HDAC enzymes are categorized into two groups: the Zn
2+-dependent enzymes, namely class I (HDAC1, 2, 3 and 8), class II (HDAC4, 5, 6, 7, 9 and 10), class IV (HDAC11), and the NAD
+-dependent enzymes (class III, also known as sirtuins)
[1][8]. Exclusive features of the HDAC8 isoform are that its gene lies in the X chromosome and that it holds key structural differences as compared to other class I HDACs. HDAC8 is smaller than HDACs 1–3 and has an independent behaviour because it lacks the C-terminal protein–protein interaction domains able to promote the formation of multiprotein complexes
[2][9]. Over the years, it has been demonstrated that HDAC8 has a lower turnover number/concentration of substrate which permits the enzyme to have a lower specificity constant (
kcat/
Km) value for the hydrolysis of acetyl lysine-containing peptides compared to HDAC1 or HDAC6, although HDAC8 presents a significant catalytic activity against tetrameric histone H3/H4 proteins
[3][10]. Other HDAC8 substrates have been identified such as p53, oestrogen-related receptor α (ERRα), AT-rich interactive domain-containing protein 1A (ARID1a), and the structural maintenance of chromosomes 3 protein (SMC3). This latter protein is the only substrate exclusively and directly deacetylated by HDAC8. Nevertheless, HDAC8 may also deacetylate other candidates by recruiting these proteins in complexes, as in the case of inv(16) fusion protein
[4][11]. It has been shown that HDAC8 hydrolyses acyl lysine residues in peptides having acyl chains of 2−16 carbons, with higher values of
kcat/
Km for the longer octanoyl, dodecanoyl-, and myristoyl-acyl chains. These long chains are able to accommodate in the binding cleft extending beyond the active site Zn
2+ cation to occupy the hydrophobic internal cavity lateral to the substrate channel. Fatty acid deacylation (removal of acyl groups with 2–16 carbons from octanoyl, dodecanoyl, and myristoyl lysine) might be a more physiologically relevant yet broad HDAC8 activity with respect to histone deacetylation, explaining how there is a small number of in vivo relevant HDAC8 substrates
[5][12].
The first HDAC8 crystal structure was described by Somoza et al. in 2004
[6][13]. In particular, the carbonyl group of the acetyl-
L-lysine coordinates with the Zn
2+ ion and forms a hydrogen bond with Y306 of human HDAC8 (
hHDAC8)
[7][14]. Furthermore, the tandem histidine pair formed by residues H142 and H143 is crucial to stabilize the transition state. H142 serves as the general base catalyst, whereas H143 serves as both the general acid and the general base catalyst (
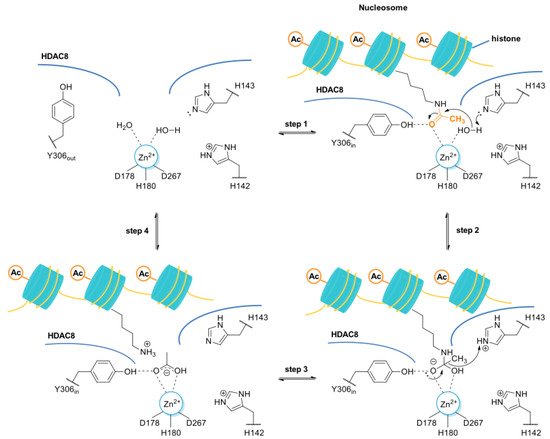
Figure 1)
[8][9][15,16].
Figure 1. Proposed deacetylation mechanism of N-acetyl lysine residue of histone protein by the HDAC8 enzyme. The residue H143, acting as a base, deprotonates a Zn2+-bound water molecule, which, in turn, performs the nucleophilic attack at the Zn2+-coordinated carbonyl group of the N-acetyl-lysine (step 1 and 2). The tetrahedral intermediate and its transition states are stabilized by the coordination bond with the Zn2+ along with the hydrogen bond interactions with Y306, H143, and H142. H143, acting as acid catalyst, assists the rearrangement of the tetrahedral intermediate to give the positively charged lysine (step 3 and 4). It was hypothesized that the side chain of Y306 may undergo conformational changes, adopting the “out” conformation in absence of the substrate, while the “in” conformation is required for the substrate binding and catalysis.
It was also observed that there is a conserved glycine-rich loop (G302GGGY) that is crucial for the maintenance of the Y306 residue. In addition, the glycine residues, especially G304 and G305, provide flexibility to the G302GGGY loop
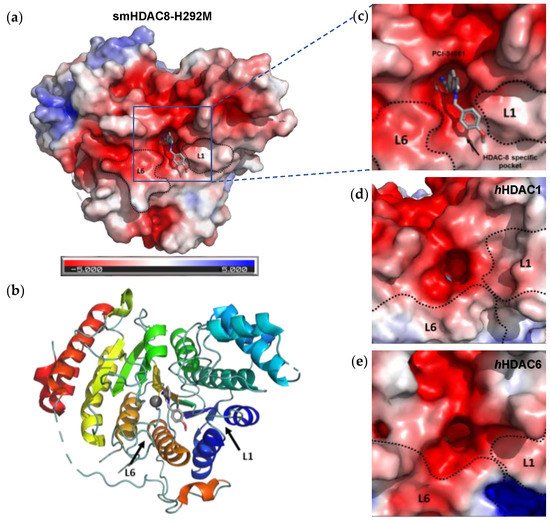
[10][17]. The active site tunnel of HDAC8 is constituted of residues such as G151, F152, H180, F208, M274 and Y306. These amino acid residues are generally hydrophobic and conserved in the class I HDACs. These amino acids help during the binding of an inhibitor containing bulky hydrophobic linker to the HDAC8 enzyme. Y306 cooperates with some aminoacidic residues of L6 and L1 loops, creating a distinctive sub-pocket, whose structural features are widely exploited in the drug design of selective HDAC8 inhibitors (HDAC8is). Compounds that assume a low energy L-shaped conformation make additional contacts with the HDAC8 catalytic domain, in comparison with other class I HDACs. In this context, PCI-34051 (
1,
Figure 2), a selective and potent HDAC8i, was extensively studied for its ability to discriminate among the other isoforms (HDAC1 IC
50 = 4 μM, HDAC6 IC
50 = 2.9 μM, and HDAC8 IC
50 = 0.01 μM), allowing for attainment of crucial information concerning the binding mode
[11][12][18,19].
Figure 2. Representation of smHDAC8-H292M in complex with 1 (PDB code: 6HSF), shown as (a) electrostatic potential surfaces, and (b) ribbon and sticks structure. Delineation of the L1 and L6 loops forming the walls of the catalytic pocket of HDAC8, HDAC1, and HDAC6: close-up representation of the electrostatic potential surfaces of (c) crystal structure of mutant smHDAC8-H292M in complex with compound 1 (PDB code: 6HSF), (d) crystal structure of hHDAC1 (PDB code: 4BXK), (e) crystal structure of hHDAC6 catalytic domain 2 (CD2) (PDB code: 5EDU). PCI-34051 (1) is depicted in light grey. Co-crystal structures were downloaded from the Protein Data Bank and visualized by Pymol (PyMOLTM 2.5.1).
However, selective HDAC8 inhibition over HDAC6 proved to be more challenging, due to the structural similarities in the active site
[13][14][20,21]. In contrast with HDAC8, L6 and L1 loops in HDAC6 isoform are able to form a larger and shallower groove that preferentially accommodates Y-shaped inhibitors endowed with extended and bulky cap groups (
Figure 2)
[15][16][22,23]. Briefly, the principal differences that define HDAC8 as a unique isoform are the following:
-
HDAC8 is an X-linked protein which acts independently, e.g., without forming any co-complexes for the activity;
-
The L1 loop of HDAC8 is closest to the enzyme active site and undergoes conformational changes, differently depending on the substrate (Figure 2);
-
L1 and L6 form a specific pocket which requires an “L” shape conformation for selective binding (Figure 2);
-
HDAC8 presents a nuclear localization sequence between the catalytic domain of the enzyme and the serine binding motif found at the end of the catalytic domain
[17][24].
1.1. HDAC8 Substrates
The typical HDAC8 histone substrates include full-length H2A/H2B, H3, and H4 histones acetylated at nonspecific lysine residues. Peptide sequences corresponding to the H4 histone tail with an acetylated lysine at position sixteen [K(ac)16] were investigated as suitable in vitro substrates
[18][25]. HDAC8 also catalyses in vitro deacetylation of the K(ac)20 site on the H4 histone tail. However, HDAC8-catalysed deacetylation of the K(ac)20 peptide is much slower than deacetylation of K(ac)16 peptides
[19][26]. Global histone acetylation levels could mask the specific deacetylation pattern of H3 by HDAC8, suggesting that non-histone targets are necessary to ascertain the exact role of HDAC8 in pathophysiological conditions. Non-histone targets are proteins whose activity could be used as a measure of HDAC activity in cells. The most important non-histone substrates identified thus far are SMC3, p53, ERRα, inv(16) and cAMP response element-binding protein (CREB). In most of these proteins, HDAC8 exhibits selectivity for certain deacetylation sites, such as arginine-Kac129 (RKac) in ERRα, RSKacFE in inv(16) fusion protein, and RHKK in p53. In general, the
N-terminal arginine at position −1 to the K(ac) and an aromatic ring (e.g., phenylalanine) at the C-terminal position +1 constitute the most effective deacetylation sites
[20][27].
1.1.1. SMC3
SMC3 is one of the most important HDAC8 substrates, with clear concentration-dependent hyperacetylation effects. Generally, SMC3 forms a clutch to hold the sister chromatids together during cell cycle progression
[21][28]. Its primary deacetylation sites are conserved K105 and K106 (human numbering). The deacetylation of SMC3 is important to segregate the sister chromatids in early mitosis and to reload the SMC3 pool for a new cell cycle. Loss of HDAC8 deacetylation activity leads to accumulation of acetylated SMC3 (Ac-SMC3) with decreased affinity towards chromatids, ultimately affecting gene transcription
[3][10].
1.1.2. p53
The tumour suppressor p53 requires HDAC8 for its expression. Depletion of HDAC8 decreases homeobox A5 (HoxA5)-dependent expression of wild-type (WT) and mutant p53 mice. The ectopic manifestation of HDAC8 increases p53 transcription while HDAC8 inactivation may be effective for p53 mutant tumour cells. Silencing or inhibition of HDAC8 mainly affects the proliferation of those cells harbouring a p53 mutation. Thus, it is reasonable to hypothesize that HDAC8is could be useful as adjuvants for the treatment of tumours carrying a p53 mutation
[3][22][10,29].
1.1.3. ERRα
Acetylation state of ERRα can be detected at four lysine residues, where post-translational modifications, as deacetylation, inhibit the DNA binding and the ERRα transcriptional activity
[23][30]. Furthermore, incubation of purified acetylated-ERR
α with HDAC8 enhances the affinity of ERR
α for DNA, which is consistent with HDAC8-catalysed deacetylation of ERRα. The acetylation site (K129(ac)) in ERRα has R121, facilitating HDAC8-mediated catalysis.
[24][31].
1.1.4. inv(16)
HDAC8 co-localizes and immunoprecipitates with smooth muscle myosin heavy chain, suggesting that HDAC8 may interact with this domain within the inv(16) fusion protein
[4][11]. When HDAC8 is overexpressed, it associates with inv(16). As a consequence, HDAC8 contributes to the transcriptional repression mediated by this chromosomal translocation fusion protein. Other HDACs do not immunoprecipitate with inv(16), suggesting that HDAC8 may be the main HDAC isoform interacting with inv(16) in vivo
[25][32].
1.1.5. CREB
HDAC8 and CREB are overexpressed in HEK293 cells, and they can be immunoprecipitated together, demonstrating that the functions of these proteins are associated. When HDAC8 is overexpressed in cells, phosphorylation of CREB decreases, which in turn inhibits CREB transcriptional activation. As HDAC overexpression can affect several targets within the cell, this inhibition may indirectly affect CREB phosphorylation. Nevertheless, CREB is not a selective partner for HDAC8 isoenzyme
[26][33].
α-tubulin (a well-known HDAC6 acetylation substrate) was also reported as a marker of HDAC8 inhibition, limiting the possibility to discriminate between HDAC6 and HDAC8 selective inhibition
[14][27][28][21,34,35]. Different studies demonstrated that the measurement of H3 acetylation levels should be the method of choice for more reliably assessing selective HDAC8 inhibition with respect to HDAC6 inhibition
[29][30][36,37].
To date, amongst the non-histone proteins, SMC3 has been acknowledged as the preferential HDAC8 acetylation substrate, while H3 represents the main histone target for HDAC8. Therefore, selective target engagement is increasingly studied by evaluating their level of acetylation, both in vitro and in vivo experiments.
2. Involvement of HDAC8 in Different Diseases
Altered HDAC8 expression has been related to different pathological conditions, generally depending on substrates (histone or non-histone) and their abnormal level of deacetylation. HDAC8 engagement can occur as a loss of enzymatic activity that requires the treatment with activators
[31][32][38,39]. Nevertheless, in most disorders, HDAC8 has an unregulated or excessive activity that can be counteracted by inhibitors. In the next sections,
thwe
researchers aanalyse the role of HDAC8 in specific diseases and, when ascertained, its mechanism of action.
2.1. X-Linked Disorders
Mutations on specific genes harboured in the X-chromosome are responsible for neurodevelopmental disorders, such as Cornelia de Lange syndrome (CdLS) and Duchenne muscular dystrophy (DMD). Several studies have enlightened that HDAC8 is enrolled in both diseases in different ways, confirming the versatile nature of this target
[20][27]. Of note, the most innovative therapeutic approaches, such as gene therapy, are not sufficient to restore the most common mutations in X-linked disorders. More in detail, the systemic nature of these pathologies requires body-wide gene transfer of non-pathogenic viral vectors, whose high dosage could cause adverse immune response
[33][34][40,41]. Thus, different strategies should be pursued to achieve an improvement in the life quality and life expectancy of affected patients.
2.1.1. Cornelia de Lange Syndrome (CdLS)
CdLS is a rare genetic disorder that leads to prenatal and postnatal growth retardation, congenital malformations, distinctive facial feature, intellectual disability
[35][42]. In general, CdLS is caused by different mutations in genes encoding proteins that regulate the cohesin complex (Nipped-B-like protein (NIPBL) and HDAC8) or its core components, (double-strand-break repair protein 21 (RAD21), SMC1A and SMC3). Mutations on the HDAC8 gene account for almost 5% of cases that display a similar phenotype, classified as CdLS 5 (OMIM#300882)
[36][37][43,44]. Patients showed a partial or complete loss of deacetylase activity, depending on the mutation localization in the gene encoding HDAC8
[35][42]. The resulting accumulation of Ac-SMC3, a bona fide HDAC8 substrate, limits the segregation of the sister chromatids and the recycling of cohesin for another cell cycle. These events lead to a cohesin-mediated transcriptional dysregulation. CdLS is mainly treated by limiting the disease progression and related symptoms
[38][45]. Medicinal chemistry efforts have been focused on the identification of HDAC8 activators to restore the residual deacetylase activity
[32][39].
A series of
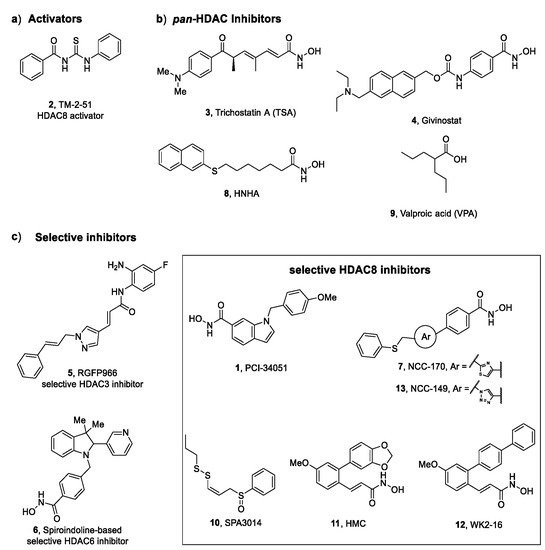
N-acetylthioureas proved effective in increasing HDAC8 activity in inhibitory enzymatic assay; thus, further studies were performed for the most potent activator, TM-2–51 (
2,
Figure 3). Singh et al. demonstrated that this compound is able to increase the catalytic turnover rate of the enzyme and to enhance HDAC8 catalysed reactions by 16-fold at 10 μM concentration, by decreasing the
Km value of the substrate
[39][46]. In addition, computational studies were performed to elucidate the pharmacodynamic profile of
2 and its mechanism of activation. The results suggested that the agonist binds to the HDAC8 enzyme in the proximity of the active site, hypothesizing the presence of an allosteric site
[39][46]. Molecular dynamic simulations proposed a “sandwich-like” binding capable of stabilizing the enzyme complex only in the presence of “loosely bound” substrates
[32][39]. The evaluation of the activity of
2 on certain CdLS HDAC8 mutants highlighted the ability of the activator to rescue the catalytic activity to wild-type levels
[40][47]. This latter finding supports the potential use of HDAC8 activators as therapeutic tools to attenuate the progression of the neurodevelopmental impairments and related deformities in CdLS patients diagnosed with HDAC8 mutation.
Figure 3. HDAC activators (2) and inhibitors (1, 3-13) explored in key pathologies: (a) thiourea-based activator of HDAC8, (b) pan-HDAC inhibitors, (c) selective HDAC inhibitors.
2.1.2. Duchenne Muscular Dystrophy (DMD)
DMD is a severe muscle-wasting disorder whose development is ascribable to mutations in the gene (DMD) encoding dystrophin that prevent the production of this protein in the striated and cardiac muscles. Despite the significant advances in insights of pathogenesis, the current therapeutic treatment is mostly aimed at relieving the symptoms. The current therapy consists of administration of corticosteroids, orthopaedic surgery, and eventually assisted ventilation
[41][42][48,49]. Different studies have enlightened the ability of
pan-HDACis to interfere with the pathogenesis of DMD. In particular, the treatment of a DMD mouse model (
mdx mice) and zebrafish model with Trichostatin A (
3, TSA,
Figure 3) ameliorated the resistance to the process of degeneration and regeneration promoted by contraction and led to the rescue of the fibre damage
[43][44][50,51]. A preliminary study performed by Consalvi et al. has revealed the beneficial effect of Givinostat (4,
Figure 3) in preventing DMD progression in mdx mice. The inhibitor exerts its action on downstream effectors of dystrophin-nitric oxide signalling, leading to a reduction of inflammation and fibrosis in muscle tissue and increasing the cross-sectional area of the myofibers
[45][52]. The study of the potential use of
4 in a long-term treatment of DMD allowed for its selection as a drug candidate for advanced phase clinical trials
[46][53]. Among the epigenetic enzymes, HDAC8 has been proven to be overexpressed in the myotubes from DMD patients and in dystrophin morpholino(
dmd-MO)-injected zebrafish embryos lacking dystrophin early in development. In the same work, the impact of HDAC8 inhibition on DMD pathogenesis was explored in a DMD zebrafish model by using PCI-34051 (
1,
Figure 3). This highly selective HDAC8 inhibitor was able to rescue the DMD phenotype by restoring skeletal muscle histomorphology and reducing inflammation in a similar manner to Givinostat (
4). Western Blot analysis was performed to evaluate the acetylation profile of a DMD zebrafish model: the results have demonstrated that the treatment with compound
1 produced an increase in α-tubulin (a marker of microtubules stability). Moreover, the assessment of cytoskeleton architecture in human DMD myoblast confirmed that HDAC8 inhibition has a crucial role in the maintenance of the skeletal muscle histomorphology
[47][54]. Taken together, these results validated HDAC8 as a potential therapeutic target to tackle DMD.
2.2. Aberrant Wound Healing
Wound healing is a transient well-orchestrated process that occurs in response to tissue damage. The consequent repair requires a controlled deposition of collagen aimed at promote scarring and tissue regeneration
[48][55]. The dysregulation of the injury-repair mechanisms can result in fibrosis, a chronic pathological condition characterized by excessive accumulation of extracellular matrix (ECM) components, such as collagen and fibronectin, that might impair the normal function of the affected tissue and relative organs
[49][56]. The fibrotic process is associated with diverse diseases that hit different organs such as lungs, kidneys, liver and heart. The causative agent is mostly unknown, even if it is possible to identify asbestos, silica, cigarette smoke, persistent infections, and chronic inflammation as possible factors able to trigger the development of uncontrolled deposition of fibrous connective material. Fibrosis initiates with the differentiation of quiescent fibroblasts, and minimally of circulating fibrocytes, into myofibroblasts. These latter have been identified as the main fibrotic effectors
[50][51][57,58]. Moreover, some studies highlighted that diverse cell types migrate to the fibrotic tissue where they are activated into myofibroblasts. In particular, epithelial cells enrich the pool of myofibroblasts since they undergo epithelial–mesenchymal transition (EMT), a transdifferentiation induced by transforming growth factor (TGF-β), predominantly through the SMAD3 signalling cascade
[52][59]. These phenotypic conversions are elicited by TGF-β1 (a well-established profibrotic mediator) together with other molecular players, comprising tumour necrosis factor-α (TNF-α), basic fibroblast growth factor (bFGF), and connective tissue growth factor (CTGF). Differently from fibroblasts, myofibroblasts stimulate excessive production of ECM components and upregulate tissue inhibitors of metalloproteinases (TIMPs) which cannot counteract the activity of ECM degradative enzymes
[53][60]. To date, treatment options are limited to cell therapy—consisting in the transplantation of bone marrow-derived mesenchymal stem cells in the lesioned tissue–and the modulation of validated targets by small-molecule drugs
[54][61]. Previous works have investigated the involvement of HDACs in fibrogenesis and how their inhibition might ameliorate the fibrotic tissue where they are overexpressed
[53][55][60,62]. It was demonstrated that many isoforms can act at specific levels, producing different pro-fibrotic effects, including the increased secretion of pro-fibrotic cytokines, growth factors
[56][63], and the induction of myofibroblast differentiation
[57][64]. HDAC8 has emerged as a valuable target for fibrosis-associated disease, and here,
thwe
researchers briefly report the scientific pieces of evidence that support this therapeutic option.
2.2.1. Pulmonary Fibrosis (PF)
PF is a rare, chronic and interstitial lung pathological condition characterized by the progressive replacement of the parenchyma with fibrotic scar tissue
[58][65]. PF leads to an impairment of gas exchange at the alveolar level with consequent dysfunctional breathing which degenerates into respiratory and heart failure. Sometimes, PF is correlated to other lung diseases such as scleroderma, sarcoidosis, and parasitic infection or can be caused by exposure to environmental agents or radiations
[59][66]. Among PF phenotypes, idiopathic pulmonary fibrosis (IPF) is the most disrupting due to its unknown aetiology and poor prognosis. The current therapy is associated with the use of nintedanib (Ofev
®, Boehringer Ingelheim, Ingelheim, Germany) and pirfenidone (Esbriet
®, Roche, Basel, Switzerland) that exert their antifibrotic effect by limiting the activation of platelet-derived growth factor receptor and decreasing the production of profibrotic growth factors and cytokines, respectively. Nevertheless, their biological effects are not sufficient to reverse the disorder; thus, new pharmacological approaches should be considered. A rising array of evidence suggests that HDACs modulation could interfere with fibrotic events in IPF
[58][65]. In particular, treatment of preclinical PF models (primary IPF fibroblasts and mouse model of bleomycin-induced lung fibrosis) with
pan-HDACis results in an attenuation of fibrotic remodelling through the epigenetic rescue of antifibrotic genes and/or the action on fibrosis-related pathways as TGF-β signalling cascade
[58][65]. Taking into consideration the chronic nature of PF, recent efforts focused on the evaluation of isoform-selective HDAC inhibition overexpressed in fibrotic lung tissue with the aim to avoid the toxic effects associated with long treatment with
pan-HDACis. A selective HDAC3 inhibitor, namely RGFP966 (
5,
Figure 3), exerts its antifibrotic effect epigenetically, by downregulating the expression of fibrogenic proteins, inflammatory cytokines and Nrf2 antioxidant enzymes
[60][67]. Furthermore, the selective spiroindoline-based HDAC6 inhibitor
6 (
Figure 3) was able to revert the fibrotic phenotype, by reducing the mRNA expression for α-SMA, collagen types I and III, fibronectin, and HDAC6 in TGF-β1-stimulated lung tissue
[16][23]. Compared to the other two isoforms, HDAC8 is mildly but significantly expressed in IPF lung tissue; thus, it was reasonable to elucidate the way in which its inhibition could impact fibrogenesis induced by TGF-β in normal human lung fibroblast (NHLFs). Saito and co-workers provided evidence that selective HDAC8 inhibition by NCC-170 (
7,
Figure 3) determines reduced contractility in TGF-β1-stimulated-NHLFs through the downregulation of α-SMA expression and cofilin dephosphorylation. Further studies suggested that HDAC8 inhibition by
7 leads to the increased H3K27 acetylation at enhancer regions of the antifibrotic mediator peroxisome proliferator-activated receptor-gamma (PPAR-γ), whose expression is promoted. Moreover, treatment with compound
7 led to a decrease in the fibrotic process in bleomycin-treated mouse lungs by repressing the expression of collagen type I and fibronectin
[61][68]. Although it was shown that selective HDAC8 inhibition could ameliorate the pathogenesis of PF, target validation requires a further biological investigation concerning the mechanism behind the antifibrotic effect in scarring lung tissue.
2.2.2. Renal Fibrosis
Renal fibrosis occurs in acute kidney injury (AKI) and chronic kidney disease (CKD), in response to renal damage
[62][69]. The fibrotic event is triggered by TGF-β1 that stimulates myofibroblasts differentiation, through the SMAD3 cascade, and promotes EMT by activation of STAT3 and β-catenin signalling pathways
[63][70]. To date, the therapeutic plan is limited to palliative care; thus, recent efforts focused on the identification of new targets to counter and delay the progression of the disease
[64][71]. The potential renoprotective and tissue reparative properties of HDACis in AKI and CKD were demonstrated in in vitro and in vivo models
[53][54][60,61]. Among
pan-HDACis, TSA (
3) was largely studied: it can inhibit fibroblast differentiation and EMT by blocking STAT3 signals in the NRK49F rat kidney fibroblast cell line. In in vivo experiments performed on diabetic rats (STZ-induced diabetic kidney),
3 is able to revert fibrotic phenotype in the kidney, as demonstrated also in NRK52E—a rat kidney epithelial cell line. Recently, the role of HDAC8 in renal fibrosis has been highlighted, suggesting its potential as a target for AKI and CKD. Long and co-workers screened a series of known selective HDAC8is, possessing different scaffolds, in the zebrafish AKI (
zfAKI) model. Notably, the treatment with PCI-34051 (
1) and two tetrahydroisoquinoline (THIQ)-based hydroxamic acids led to phenotypic
zfAKI efficacy at 4 μM. The compounds were also evaluated in human kidney organoids that can simulate some of the fibrotic events arising after AKI: among the three inhibitors,
1 exhibited an interesting ability in reducing collagen deposition. Encouraged by these results, compound
1 was also tested in a mouse model of AKI, showing a reduction of mRNA markers of renal fibrosis with no direct effect on the disorder
[65][72]. Following these latter findings, previous work evidenced that HDAC8 inhibition with
1 could modulate the expression of profibrotic markers, attenuating renal fibrosis induced by unilateral ureteral obstruction in vivo and TGF-β1 exposure in vitro. The
res
earchtudy allowed for demonstrating HDAC8 expression in renal tubular epithelial cells and its upregulation in a murine model, after UUO injury. As a result, the increasing deacetylase activity on cortactin, a non-histone HDAC8 substrate, implies overexpression of α-SMA, collagen type I, and fibronectin that can be suppressed by treatment with
1. Moreover, HDAC8 inhibition mediates the dephosphorylation of SMAD3, STAT3, and β-catenin, which translates into the activation of the TGF-β1-related signalling pathways underlying fibrogenesis. PCI-34051 exerts its antifibrotic activity in renal epithelial cells either through the arrest of the cell cycle at the G2/M phase and the upregulation of the transcription factor Snail, two events that prevent EMT. As reported in the UUO kidney, selective HDAC8 inhibition re-established the level of expression of Klotho and BMP-7, two major reno-protective proteins
[66][73].
Taken together, these results pinpoint HDAC8 inhibition as a new potential strategy to yield important beneficial effects in renal fibrotic diseases.
2.2.3. Liver Fibrosis
Liver fibrogenesis is an aberrant response to hepatic damage associated with diverse pathological conditions such as hepatitis virus infection, alcoholic and non-alcoholic fatty liver diseases. The key cellular mediators are the hepatic stellate cells which undergo phenotypic conversion into myofibroblasts after the activation of several profibrotic pathways, predominantly arbitrated by TGF-β. Once activated, they drive the fibrotic process through the overproduction of ECM proteins which is, in turn, enhanced by their irregular degradation from the metalloproteinases (MMPs). The effective pharmacological approach aimed to arrest the progression of liver fibrosis remains limited to the eradication of the main causative agent, while it is tempting to direct the research towards the development of new antifibrotic agents, regardless of their aetiology
[54][61]. Given the considerable involvement of HDACs in fibrosis, their inhibition appeared to be a valuable strategy to reduce liver fibrosis
[67][74]. Based on these observations, Park and co-workers identified HNHA (
8,
Figure 3), a
pan-HDACi, as an antifibrotic modulator in hepatic tissue. The compound inhibits the activation and proliferation of PDGF-induced mouse primary HSCs and ameliorates liver fibrosis caused by bile duct ligation (BDL) in rats. It can exert multiple actions on different steps of fibrotic event, including the expression of profibrogenic effectors (α-SMA, TGF-β, collagen type I)
[68][75]. Moreover, it was demonstrated that valproic acid (
9, VPA,
Figure 3), another
pan-HDACi, prevents the activation of thioacetamide-treated-HSCs by promoting apoptosis and contributes to reducing the accumulation of ECM proteins via the enhancement of MMPs expression
[69][76]. Furthermore, a deeper investigation led to confirm the epigenetic effect of VPA inhibition consisting additionally in modulating the expression of the miRNAs responsible of fibrogenesis
[70][77]. Although there is much evidence that supports the beneficial effect of
pan-HDACis in liver fibrosis, the research community moved on the identification of inhibitors endowed with a remarkable selectivity towards a specific isoform. Considering that HDAC8 is overexpressed in BDL mice, it was reasonable to hypothesize its correlation with hepatic fibrogenesis and, in turn, to verify it in cholestatic liver injury. The treatment of BDL mice with the selective HDAC8i SPA3014 (
10,
Figure 3) produced a diminished expression of collagen type I and α-SMA which derives from the induced suppression of TGF-β expression. These biochemical effects were supported by gross and histopathologic analysis that confirmed the capability of the HDAC8i to limit the shifting towards the fibrotic phenotype. In conformity with these in vivo data, SPA3014 decreases TGF-β1 expression and related downstream signalling pathways—MAPK-Smad2/3 and JAK2-STAT3—in LX-2
hHSCs, exerting a hepatoprotective effect. Furthermore, HDAC8 inhibition results in the upregulation of PPAR-γ which contributes to attaining the antifibrotic effect
[71][78]. The target validation of HDAC8 for the treatment of liver fibrosis represents a considerable starting point to develop new agents whose therapeutic use can be declined in diverse chronic liver diseases.
2.2.4. Cardiac Fibrosis
Cardiac fibrosis is a pathological condition in which the main heart functions are compromised. In particular, atrial interstitial fibrosis affects the generation and conduction of electric signals from the sinoatrial node, driving the development of arrhythmia and thrombosis. Although mostly concomitant with cardiac hypertrophy, ventricular fibrosis is responsible for tissue stiffness and diastolic dysfunctionality
[54][61]. In this context, HDACis displayed an interesting antifibrotic activity equally in atrial and ventricular fibrosis
[72][79]. More in detail, TSA (
3) revealed to be effective in attenuating atrial fibrosis and fibrillation when tested in HopX transgenic mice with left ventricular hypertrophy
[73][80]. Another example concerns the use of VPA (
9) to treat hypertensive rats. Compound
9 prevented cardiac hypertrophy and fibrosis by regulating the acetylation of mineralocorticoid receptors
[74][81]. Several works that tried to elucidate the mechanism of action of
pan-HDACis are coupled to as many attempts to clarify the contribution of the isoform-selective inhibition to the final antifibrotic effect. Recently, the role of HDAC8 was evaluated in cardiac hypertrophy and fibrosis using isoproterenol-induced cardiac hypertrophy model where the expression of HDAC8 is upregulated. The authors provided evidence that the mRNA levels of the fibrosis markers collagen type I, fibronectin, and CTGF diminish in response to the treatment with
1, as well as the expression of α-SMA and TGF-β1 mRNA and related proteins. The mechanism by which the compound exerts its antifibrotic effects implies the inactivation of p38/MAPK, a downstream HDAC8 target involved in the onset and progression of cardiac fibrosis
[75][82]. The inhibitor
1 was also selected to evaluate in which measure HDAC8 inhibition could impact transverse aortic constriction (TAC)-induced heart failure in mice. Western blotting analyses and q-RT-PCR have enlightened that the small molecule behaves as a negative regulator of fibrosis-related genes such as
COL1A1, FN1, ACTA2, and
TGFΒ1 in TAC mice. Further examination has indicated that compound
1 can reverse the upregulation of TGF-β1 and phosphorylated Smad2/3, central key actors of fibrotic signalling, in vivo and in vitro TGF-β1-stimulated cardiac fibroblasts. The selective inhibition of HDAC8 mitigates cardiac fibrosis in two different disease models via overlapping mechanisms. These results allow for identifying HDAC8 as a potential pharmacological tool for the treatment of cardiac disorders accompanied by fibrotic state
[76][83].
2.2.5. Aberrant Wound Healing Associated with Diabetic Foot Ulcers (DFU)
Concomitantly with tissue remodeling, angiogenesis is a well-established process that occurs during wound healing, in the growing tissue. About 4% of diabetes mellitus patients present diabetic foot ulcers, multifactorial vascular complications that require lower limb amputation in the most severe cases References
[77][78][79][84,85,86]. Many efforts have been focused on the identification of efficacious pharmacological approaches to treat different kinds of DFU that share impaired angiogenesis. In this frame, a recent work identified the LINC01435/YY1/HDAC8 pathway as a promising target for its impact on the angiogenetic process in diabetic wounds
[80][87]. A preliminary study was performed to verify the effect induced by exosomes from high glucose-pretreated immortalized human epidermal cells (HG-Exos) in diabetic mice, whose wound repair improved after the treatment. Further investigations allowed for attributing this effect to exo-mediated uptake of LINC01435—A long noncoding RNA—that suppresses the migration and tube formation of human umbilical vein endothelial cells (HUVECs). Considering the role of the Notch pathway in angiogenesis, the authors extended the inspection of potential antiangiogenetic targets to various players engaged in Notch signalling, including the HDACs family
[81][82][83][88,89,90]. This analysis highlighted the overexpression of HDAC8 in HUVECs, promoted by the nuclear translocation of the transcription factor YY1. Additional experiments highlighted that HDAC8 knockdown could enhance vessel formation and migration of HUVECs, enforcing the evidence that HDAC8/Notch signalling is involved in the antiangiogenetic effect of Exo-LINC01435. These promising results should encourage the research to exploit HDAC8 selective inhibition as a potential approach to promote wound healing in DFU.
2.3. Cancer
The term “cancer” encompasses a group of multifactorial diseases characterized by abnormal cell proliferation of the affected cells which might develop the capacity to invade other nearby sites or diffuse in other organs, generating metastatic tumours. Oncogenesis can occur via the activation of multiple pathways with the involvement of several targets, whose pharmacological regulation constitutes one of the most effective approaches against this disorder. Worldwide, metastatic cancers represent the second leading cause of death. Despite the considerable arsenal of available drugs, there is an urgent need for novel therapeutics due to their severe side effects often accompanied by drug resistance phenomena. Given that epigenetic modifications can contribute to the onset and progression of malignancies, the potential contribution of HDACs has been largely explored in different cancer types, leading to the FDA approval of
pan-HDACis as anticancer drugs
[84][91]. In this context, the search for new inhibitors endowed with enhanced isoform-selectivity appears more attractive and challenging, since this strategy might overcome the undesired effects deriving from treatment with broad-spectrum HDAC inhibitors. Among the isoforms under investigation, HDAC8 has been proven to interfere with tumorigenesis. Given that HDAC8 plays a pivotal role for p53 expression, its enzymatic inhibition showed to be effective in tumour cells harbouring a p53 mutation. Nevertheless, further studies proved that HDAC8is can exert their antitumoural action through the activation of alternative mechanisms. Several reviews elaborated on the beneficial effect deriving from HDAC8 inhibition in a wide range of cancers taking place in blood, breast, liver, colon, lungs, and nervous system
[3][20][85][86][10,27,92,93]. In this section,
thwe
researchers give an overview of the most recent advances relative to HDAC8 engagement in some of the mentioned cancer types clustered in haematological and solid tumours.
2.3.1. Haematological Malignancies
Acute myeloid leukaemia (AML) can be classified as a heterogeneous series of malignant disorders featuring aberrant proliferation and differentiation of myeloid cell lines in the bone marrow. Based on the early discovered antiproliferative properties of HDAC8is against malignancies, further investigations were carried out to deeply comprehend their mechanism of action. Recently, Spreafico and co-workers demonstrated that treatment of AML cell lines (OCI-AML5, PLB985, THP-1, and AML193) with
1 restrained cell proliferation, inducing the cell cycle arrest along with apoptosis via activation of p53 signalling. This result was comparable with those obtained from the experiment conducted in the zebrafish disease model overexpressing HDAC8. Since p53-null HL60 is also sensitive to HDAC8 inhibition by
1 at 50 μM, some efforts were requested to identify the alternative mechanism underlying the cycle arrest in G0/G1 phase. The outcomes of the experiments indicated that HDAC8 inhibition determined the activation of the canonical Wnt pathway, whose dysregulation is associated with AML
[87][94].
The potential involvement of HDAC8 was also explored in mantle cell lymphoma (MCL), a severe non-Hodgkin lymphoma that requires the use of high-dosed antitumour drugs with a negative impact on the therapeutic index. The selective inhibition of HDAC8 can induce caspase-dependent apoptosis in MCL cell line resulting in a cytostatic and cytotoxic effect. In contrast to
pan-HDACis, inhibition induced by compound
1 did not impair the viability of natural killer cells but preserved their functional response to cancer conditions
[88][95].
2.3.2. Solid Tumours
A growing body of work has pinpointed epigenetic modulation as a valuable strategy to tackle breast cancer. In the research of new epigenetic targets, HDAC8 exhibited antiproliferative properties in different breast cancer subtypes. Chiu et al. provided evidence that HMC (
11,
Figure 3), a selective HDAC8i, was able to cause caspase-dependent apoptosis in the MCF-7 cell line through the suppression of Akt/mTOR signalling and the activation of PPAR-γ pathway, coupled with DNA damage induced by ROS production. In addition,
11 stimulated autophagy, an explicit survival mechanism that enhances cell growth inhibition in the treated MCF-7 cells
[30][37]. Further investigations have enlightened that HDAC8 promoted dissemination of breast cancer cells in vitro and in vivo, through the activation of EMT, a central process underlying metastasis. Enzymatic inhibition with
1 preserved protein stability of Snail, a fundamental modulator of EMT, via other two concomitant and correlated mechanisms: the increase in GSK-3β phosphorylation which is, in turn, assisted by HDAC8-regulated AKT phosphorylation. This finding represents a good starting point to further elaborate on the therapeutic effect of HDAC8i for severe and metastasized breast cancer
[89][96].
Consistent with this latter work, it was proven that treatment with
1, as monotherapy or in combination with cyclophosphamide, adriamycin, and 5-fluorouracil (CAF), impaired tumour cell survival in basal-like breast cancer, by suppressing some transcription factors involved in the EMT process (Gata3, Elf5, Rora and Grhl2). The effect was evaluated in murine and human basal-like breast cancer cell lines—pG-2 and HCC1806, respectively
[86][93]. Experiments conducted in HeLa cervical cancer cells could attribute the antiproliferative effect to the downregulation of HDAC8 activity. Treatment with inhibitor
1 decreased the level of the acetylated α-tubulin (Ac-Tub), which plays an important role in cell migration and in G2/M phase of the cell cycle. As a final effect, reduction of mitosis and a diminished capacity of dissemination were observed
[90][97].
Many examples in the literature reported the use of HDACis to empower the immune checkpoint blockade in cancer by restoring or enhancing the capability of T-cells in detecting and destroying the tumour cells. Furthermore, the immunomodulatory properties of HDACis were exploited to potentiate the action of immune checkpoint inhibitors, leading to a synergistic antitumour effect
[91][92][93][94][95][96][97][98][99][98,99,100,101,102,103,104,105,106].
Recently, it was discovered that acetylation of H3K27, induced by selective HDAC8 inhibition, entails the secretion of T cell–recruiting chemokines by hepatocellular carcinoma cells, resulting in enhanced CD8
+ T cells infiltration in preclinical disease model
[100][107].
Furthermore, it was found that the selective HDAC8i
1 could impair the viability and the migration of human and murine glioma cells, and in agreement with these findings, it was proven to reduce the tumour size in mice models of glioma. The effect is ascribable to the increase in the Ac-Tub, as seen in HeLa cells. In addition, HDAC8 inhibition enforces the immune response mediated by natural killer cells through positive regulation of the transcription of ligands for NKG2D receptor, whose activation is responsible of NK cytotoxicity in tumour cells
[101][108]. The anticancer properties of
1 have been exploited in glioblastoma multiforme (GBM), with the aim to overcome the adverse effect of the chemotherapy with temozolomide (TMZ). This drug induces an increase in O
6-methyl-guanine DNA methyltransferase (MGMT) which acts in the repair of DNA damage. Compound
1 allowed reversing this drug response in TMZ-resistant cells, by cooperating with ADRM1, the proteasome receptor. Therefore, HDAC8 inhibition determines reduced viability in GMB cell line due to the arrest of the cell cycle, enhanced by a failed repair of DNA damage
[102][109].
Neuroblastoma is a typical childhood tumour of the peripheral nervous system which has drawn the attention of the research community because of its high lethality rate. Among the screened targets, in vitro and in in vivo studies revealed that HDAC8 is involved in this pathology. HDAC8 inhibition is correlated to the block of cell proliferation, the induction of cell cycle arrest and the promotion of differentiation in neuroblastoma cell line
[103][110]. Recently, further studies have strengthened the idea that HDAC8is could represent potential therapeutic tools to reverse the disorder, especially when their action is potentiated by other factors, which are discussed in the next paragraphs
[104][105][111,112].