Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | George Saitakis | -- | 4142 | 2022-09-17 10:36:05 | | | |
| 2 | Vivi Li | Meta information modification | 4142 | 2022-09-19 03:24:14 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Saitakis, G.; Chwalisz, B.K. Typical and Atypical Optic Neuritis. Encyclopedia. Available online: https://encyclopedia.pub/entry/27270 (accessed on 23 July 2026).
Saitakis G, Chwalisz BK. Typical and Atypical Optic Neuritis. Encyclopedia. Available at: https://encyclopedia.pub/entry/27270. Accessed July 23, 2026.
Saitakis, George, Bart K. Chwalisz. "Typical and Atypical Optic Neuritis" Encyclopedia, https://encyclopedia.pub/entry/27270 (accessed July 23, 2026).
Saitakis, G., & Chwalisz, B.K. (2022, September 17). Typical and Atypical Optic Neuritis. In Encyclopedia. https://encyclopedia.pub/entry/27270
Saitakis, George and Bart K. Chwalisz. "Typical and Atypical Optic Neuritis." Encyclopedia. Web. 17 September, 2022.
Copy Citation
Optic neuritis (ON) is an inflammatory condition involving the optic nerve. Several important typical and atypical ON variants are now recognized. Typical ON has a more favorable prognosis; it can be idiopathic or represent an early manifestation of demyelinating diseases, mostly multiple sclerosis (MS). The atypical spectrum includes entities such as antibody-driven ON associated with neuromyelitis optica spectrum disorder (NMOSD) and myelin oligodendrocyte glycoprotein antibody disease (MOGAD), chronic/relapsing inflammatory optic neuropathy (CRION), and sarcoidosis-associated ON. Appropriate and timely diagnosis is essential to rapidly decide on the appropriate treatment, maximize visual recovery, and minimize recurrences.
atypical optic neuritis treatment
typical optic neuritis
MOG
NMO
MS prevention
1. Introduction
Optic neuritis (ON) is an inflammatory condition involving the optic nerve but is far from being a uniform condition, and several important variants are now recognized that can be stratified into typical and atypical forms. Typical, ON usually manifests in young adults, especially women, between 18 and 45 years of age, and can be idiopathic or represent an early manifestation of demyelinating diseases, mostly multiple sclerosis (MS). The atypical spectrum includes entities such as neuromyelitis optica spectrum disorder (NMOSD), myelin oligodendrocyte glycoprotein antibody disease (MOGAD), chronic/relapsing inflammatory optic neuropathy (CRION), and sarcoidosis-associated ON, and in all of these, the clinical presentation, visual prognosis, and recurrence risk differ from typical ON. Importantly, optimal treatment approaches are also not uniform, making it essential to more accurately differentiate these entities based not only on their clinical presentation but also their pathogenesis.
2. Typical Optic Neuritis
Optic neuritis (ON) is an inflammatory condition involving the optic nerve, manifested usually in young adults, especially women, between 18 and 45 years of age [1]. The majority of the cases are idiopathic, but ON can be associated with demyelinating diseases, most commonly multiple sclerosis (MS). Optic neuritis represents one of the most frequent phenotypes of MS relapse, and occurs as the first demyelinating event in about one out of three MS patients [2]. MS is characterized by the presence of plaques that form in the CNS in combination with inflammation, demyelination, axonal injury, and axonal loss. The plaques are located primarily in the white matter of the brain, spinal cord, and optic pathways, but there is also involvement in the gray matter [3][4]. Depending on their stage of development, they contain varying proportions of immune cells and immunoreactive substances [5]. Plaques are expressed in all forms of MS, but vary over time quantitatively and qualitatively, showing a profound heterogeneity in the structure and immunopathological patterns of demyelination and oligodendrocyte pathology between relapsing-remitting and progressive forms of MS [6]. MS likely represents a T-cell-mediated autoimmune disorder with a predominance of CD8+ cells. The dominant theory is that inflammatory lesions in MS consist mainly of CD8+ and CD4+ T cells, and activated microglia and macrophages [7][8]. There is evidence regarding the suppression of functions that restricts CD4+ T-cell responses, and the tissue-damaging role of CD8+ T cells is reported to co-localize with axonal pathology [9][10]. Experiments in humanized transgenic mice showed that the specific interaction of CD8+ T cells with target cells requires MHC-I expression, which is tightly regulated in neurons and MHC-I molecules, only in response to danger signals such as pro-inflammatory cytokines IFN-γ or TNF-α [9]. However, the role of B cells has also become apparent, as evidenced, for instance, by the effectiveness of B cell inhibition as an MS disease-modifying therapy (DMT).
3. Pathophysiology of ON
In the acute phase, ON pathology is characterized by optic nerve abnormalities and inflammatory demyelination. More specifically, predominant T cell, B cell, and glial cell activation within the nerve increases pro-inflammatory cytokines, leading to the activation of microglia and monocyte-derived macrophages, and further recruitment of CD4− and CD8+ T cells [11]. The subsequent inflammation leads to demyelination, reactive gliosis, and axonal death [12]. Pro-inflammatory cytokines and cytotoxic factors target myelin-producing oligodendrocytes (OLGs) and oligodendrocyte precursor cells (OPCs), causing apoptosis and exacerbating axonal demyelination [13][14][15][16]. Mature OLGs that survive demyelination are unable to produce new myelin sheaths. Remyelination, therefore, requires the migration and regeneration of oligodendrocytes from OPCs [17].
It is worth noting that the acute inflammatory lesions of the afferent visual pathway cause retrograde degeneration of retinal ganglion cells (RGCs). It has been demonstrated that RGC loss is associated with a reduction in post-synaptic proteins and neurite projections, and with persistent microglia and astroglia activation in the inner retina with high levels of iNOS (inducible nitric oxide synthase), IL (interleukin)-1α, TNF (tumor necrosis factor)-α, and C1q (complement component 1q) [15]. Thus, the development of therapeutic agents should focus on anti-inflammatory, anti-apoptotic, and remyelinating mechanisms to achieve neuroprotection and neuro-regeneration in the optic nerve and retina.
4. Acute Treatment of Typical Optic Neuritis/Clinically Isolated Syndrome
In general, MS is characterized by its tendency for recurrence in proximity to a previously affected site, as has been observed radiologically [18][19] and confirmed in post-mortem pathological studies [20]. Lotan et al. showed that in MS, recurrent episodes of ON tend to attack the same optic nerve that was affected before [21]. Similar findings come from a 2011 study [22]. Potential explanations for the recurrent nature of ON in MS is the disruption of the blood–brain barrier during the initial insult and antigenic change and expansion, leading to epitope spreading as a pathogenic event leading to a chronic CNS demyelinating disease [23].
Based on the presence of prominent immunologic activity in the pathologic samples of MS patients and oligoclonal bands in the CSF of most MS patients, it has been suggested that the disease is an immune-mediated disorder [6][24][25][26]. However, there are alternative theories claiming that MS is not a homogenous condition, thus not fulfilling the criteria of an autoimmune disease [27][28][29]. Much effort has been invested in identifying the autoantigen(s) against which the oligoclonal bands are directed, so far without success. It is believed that the inflammatory attack is not an outcome of an immune response directed against a specific auto-antigen. Thus, in MS, unlike NMOSD and MOG antibody disease, the immune response may be nonspecific and triggered by tissue changes induced by the previous attack.
Corticosteroid use has traditionally been the common approach for the treatment of ON, with the first implementation dating back to the 1950s [30]. Data from the United States demonstrate that the majority of ophthalmologists and neurologists in the 1980s used to treat their patients with optic neuritis with standard oral doses of corticosteroids, despite the lack of convincing evidence of efficacy [30][31]. The Optic Neuritis Treatment Trial (ONTT) was the first multicenter, randomized, collaborative clinical trial of ON [30][31]. Fifteen centers in the United States participated in the ONTT, recruiting 457 patients between 1 July 1988 and 30 June 1991. Patients were enrolled who had acute unilateral optic neuritis with visual symptoms lasting 8 days or less, aged between 18 and 45 years, with no previous history of optic neuritis in the affected eye, no evidence of associated systemic disease other than MS, and no previous treatment with corticosteroids for MS or optic neuritis [31]. The mean age of patients at study entry was 32 years, 77% of patients were women, and 85% identified as white. The participants were randomized either to be treated with oral prednisone (1 mg/kg daily for 14 days), intravenous methylprednisolone (250 mg every 6 h for 3 days) followed by oral prednisone (1 mg/kg daily for 11 days), or oral placebo. Each regimen was followed by a short oral dosage taper consisting of 20 mg of prednisone (or placebo) on day 15 and 10 mg of prednisone (or placebo) on days 16 and 18 [29][30]. In general, steroid treatment was well tolerated, with only minor adverse effects (sleep disturbance, mild mood change, upset stomach, facial flushing, mild weight gain), except for a case of acute transient depression and another patient that suffered from acute pancreatitis. Patients were evaluated in seven follow-up visits during the first 6 months, at 1 year, then yearly through 1997, in 2001 through 2002, and finally in 2006. According to the study design, the primary outcome for the treatment group comparison was set at 6 months.
The study findings demonstrated that the natural course of visual functions after an episode of typical optic neuritis, either treated or untreated, is one of a rapid visual recovery beginning within 2 weeks after the onset of symptoms, with most of the recovery often taking place after 4 to 6 weeks, and further slow recovery over several months, even up to 1 year [2]. In almost all patients, regardless of the treatment group and initial severity of visual losses, some improvement began within the first 30 days [2][30]. Of clinical relevance, recurrences of optic neuritis occurred more commonly in patients treated with oral prednisolone alone; within 2 years from diagnosis, the probability of recurrence in either eye was almost 2-fold higher in the low-dose prednisone group (30%) than in either the placebo group (14%) or the high-dose intravenous group (16%) [30][31][32][33]. The ONTT showed that vision recovered faster in the intravenous group than in the other groups, although the difference among the three groups had faded by 30 days. However, at 6 months, qualitative features such as contrast sensitivity, visual field, and color vision were still slightly better in the intravenous group. By contrast, the prednisone group compared with the placebo group demonstrated no significant differences in the rate of recovery or the 6-month outcome for any aspect of the visual function. At the 6-month point, patients in all three treatment groups had a median visual acuity of 20/16, and fewer than 1 out of 10 patients had a visual outcome of 20/50 or worse. At the 1-year follow-up, there was no statistically significant difference in visual function among the groups. Visual acuity was 20/40 or better in 95% of the placebo group, 94% of the intravenous steroid group, and 91% of the oral steroid group at 1 year. After 15 years, 72% of the eyes affected with optic neuritis had visual acuity of ≥20/20, and 66% of the patients had ≥20/20 acuity in both eyes [1]. A 2015 Cochrane Systematic Review also reported the failure of intravenous steroids to improve vision outcomes in ON [34]. The ONTT also found that among the 389 patients without a diagnosis of clinically probable or definite MS at study entry, the intravenous steroid group showed a lower rate of development of clinically definite MS within the first 2 years (7.5%) than did the placebo (16.7%) or prednisone (14.7%) groups, but this apparent protective effect was not sustained at 3 years [30]. By 5 years, the treatment had no significant effect on the development of MS. Most of the aforementioned intravenous treatment group benefits on the development of MS were observed in patients with brain findings on the magnetic resonance imaging (MRI) at baseline, because the rate of MS among patients without baseline MRI lesions was so low that therapeutic efficacy could not be determined.
Some potential limitations of the trial include the definition of symptom onset (timed from the visual loss but not from the onset of pain), inclusion of possible MOG cases, the validity of the primary outcome measure of high-contrast visual acuity, and the lack of pharmacokinetic data (making it difficult to develop a plausible biological explanation for as to why oral vs. intravenous corticosteroids should be harmful compared with intravenous corticosteroids). In addition, the long interval between the onset of symptoms and initiation of treatment in ONTT (up to 8 days) leaves open the possibility that a “critical time window” may have been missed, and that more vision loss could be prevented if treatment was initiated in the early inflammatory phase (within 48 h) [35][36]. Experimental evidence supports such a critical time window for treatment initiation in optic neuritis, as it has been shown that inflammation of the optic nerve precedes demyelination and axonal degeneration by about 2 days, and irreversible damage to the axonal cytoskeleton occurs within 5–7 days [35][37]. Indeed, a retrospective study demonstrates significant improvement in both functional and structural outcomes in patients with relapsing ON when treatment is initiated early [38].
The current standard of care for typical optic neuritis, still based on the results of the ONTT, is either no treatment in mild cases or the administration of intravenous steroids to accelerate visual recovery [30][39][40]. A proton pump inhibitor may also be given to prevent peptic ulcers. There is no role for low-dose oral prednisone [31]. This reasoning is consistent with a Cochrane meta-analysis as well [41].
Since the publication of the ONTT, other studies have shown that high-dose oral corticosteroids and high-dose IV methylprednisolone are bioequivalent, and have similar effects on MRI outcomes and clinical MS relapse [2]. Morrow et al., in 2018, showed in a single-blind randomized clinical trial that the efficacy of high-dose oral steroids is bioequivalent to and shows no inferiority to intravenous steroids. More specifically, 55 participants were randomized to either methylprednisolone sodium succinate (1000 mg, IV) daily for 3 days or oral prednisone (1250 mg) daily for 3 days. Improvements in vision were noticed at 1 month and at 6 months [2]. Compliance with this oral regimen has been previously shown to be very high [2]. Similar results were cited by the COPOUSEP trial in France [42]. In addition, a Cochrane review in 2008 compared the efficacy of the two forms of steroid administration and found them to be equally effective. Studies have also shown that intravenous dexamethasone in a dose of 200 mg/day had comparable efficacy to 1 g/day of intravenous methylprednisolone, and has the advantage of low costs and fewer side effects [39]. Intramuscular or subcutaneous adrenocorticotropic hormones are also approved for the treatment of ON- and MS-related relapses [40].
Intravenous immunoglobulin (IVIg) has a potential role in the management of acute optic neuritis, though evidence is limited, and the agent is typically reserved for the treatment of patients with steroid-refractory ON. IVIg may cause rash, fever, and, rarely, aseptic meningitis, thrombosis, hemolysis, and renal dysfunction [39]. In general, plasma exchange (PLEX) is typically favored over IVIg to manage MS relapses that are not responsive to steroid treatments. PLEX is associated with a number of potential side effects including myocardial infarction, arrhythmia, hemolysis, central line placement risk, and death in a small percentage of patients [43]. More recently, high-dose cyclophosphamide (50 mg/kg per day for 4 consecutive days, followed by a granulocyte-colony-stimulating factor 6 days after completion) was evaluated in nine patients with aggressive RRMS as a rescue treatment for acute fulminant relapses. Potential side effects of the short-term high-dose cyclophosphamide monotherapy in patients with MS include neutropenia and infection [40][44].
5. Long-Term Treatment: Immune Prophylaxis against Optic Neuritis Relapses/Progression to Multiple Sclerosis
5.1. Mechanisms of Action in Interferon β in MS and Optic Neuritis
Interferons (IFNs) have been recruited as a potential therapeutic option for MS based on their immunomodulatory and antiproliferative properties [45]. It is believed that IFNs act via several overlapping mechanisms such as the down-regulation of the major histocompatibility complex (MHC) class II expression present on the antigen-presenting cells, the induction of T-cell production of interleukin 10 (IL-10), and thus a shift in the balance toward anti-inflammatory T helper (Th)-2 cells, and the inhibition of T-cell migration as a result of a blockade of metalloproteases and adhesion molecules [46] (Figure 1: a synopsis of IFN mechanisms of action).
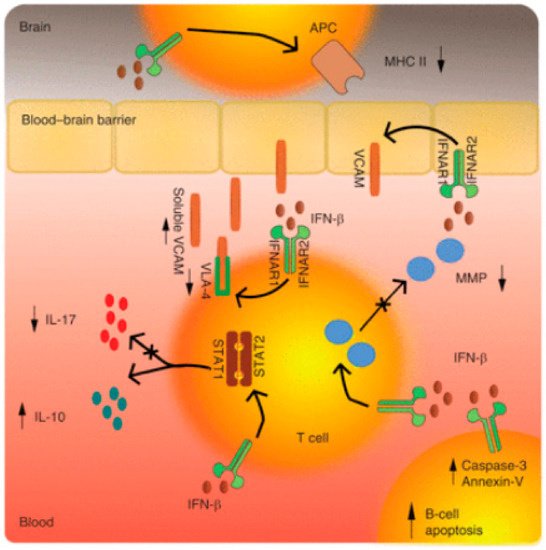

Figure 1. Molecular Mechanisms of Action of Interferon β [45].
The actions of IFNs are mediated through transcriptional factors and subsequent gene regulation. The major route in which IFN-β produces its effect is by activating the Janus kinase (JAK) signal transducers and activators of the transcription (STAT) pathway. More specifically, IFN-β binding to the type I IFN receptor causes phosphorylation of STAT1 and STAT2 and the formation of STAT1-STAT2 heterodimers, which translocate to the nucleus, bind the IFN-stimulated response element (ISRE), and modulate the expression of ISRE-regulated genes [47]. It has been demonstrated that the cellular response to IFNs is complex and results in changes in the expression of more than 500 genes representing ∼0.5% of the human genome [48]. Rizzo et al., focusing on the pivotal role of B cells in MS immunopathology, investigated the mechanism of B-cell apoptosis. The up-regulation of mechanisms that require FAS-receptor/TACI (transmembrane activator and CAML interactor) signaling and the production of apoptotic markers such as Annexin-V and caspase-3 were shown as specific inducers of B-cell apoptosis [49].
5.2. Glatiramer Acetate (GA)
The mechanism of action of GA has long been an enigma. GA has well-established immunomodulatory properties, promoting the expansion of anti-inflammatory and regulatory Th2 and Treg cells and inducing the release of neurotrophic factors. Using various genetically modified mouse strains, as well as human monocytes, Molnarfi et al. showed that GA inhibited the TRIF-dependent pathway, resulting in a reduction in IFN-β production [50] (Figure 2). This observation is consistent with the earlier demonstration that STAT1 phosphorylation is reduced upon activation in type II monocytes [51]. These findings provide a key anti-inflammatory mechanism connecting innate and adaptive immune modulation in GA therapy. Animal studies have also shown that GA-reactive Th2 cells migrate to the CNS and accumulate at the site of active lesions. Thus, GA-reactive T cells provide the effector arm in treatment. However, GA treatment influences both innate and adaptive immune compartments, and it is now recognized that antigen-presenting cells (APCs) are the initial cellular targets for GA, and it is the modulation of the APC compartment to anti-inflammatory (M2) phenotypes that leads to an expansion in regulatory Th2 and Treg cells. In addition, the anti-inflammatory (M2) APCs induced following treatment with GA are responsible for the induction of anti-inflammatory T cells that contribute to its therapeutic benefit [52]. Mechanisms of action of GA that promote immunomodulation and neuroprotection are not mutually exclusive, and several may contribute to the efficacy of the drug (Figure 3).
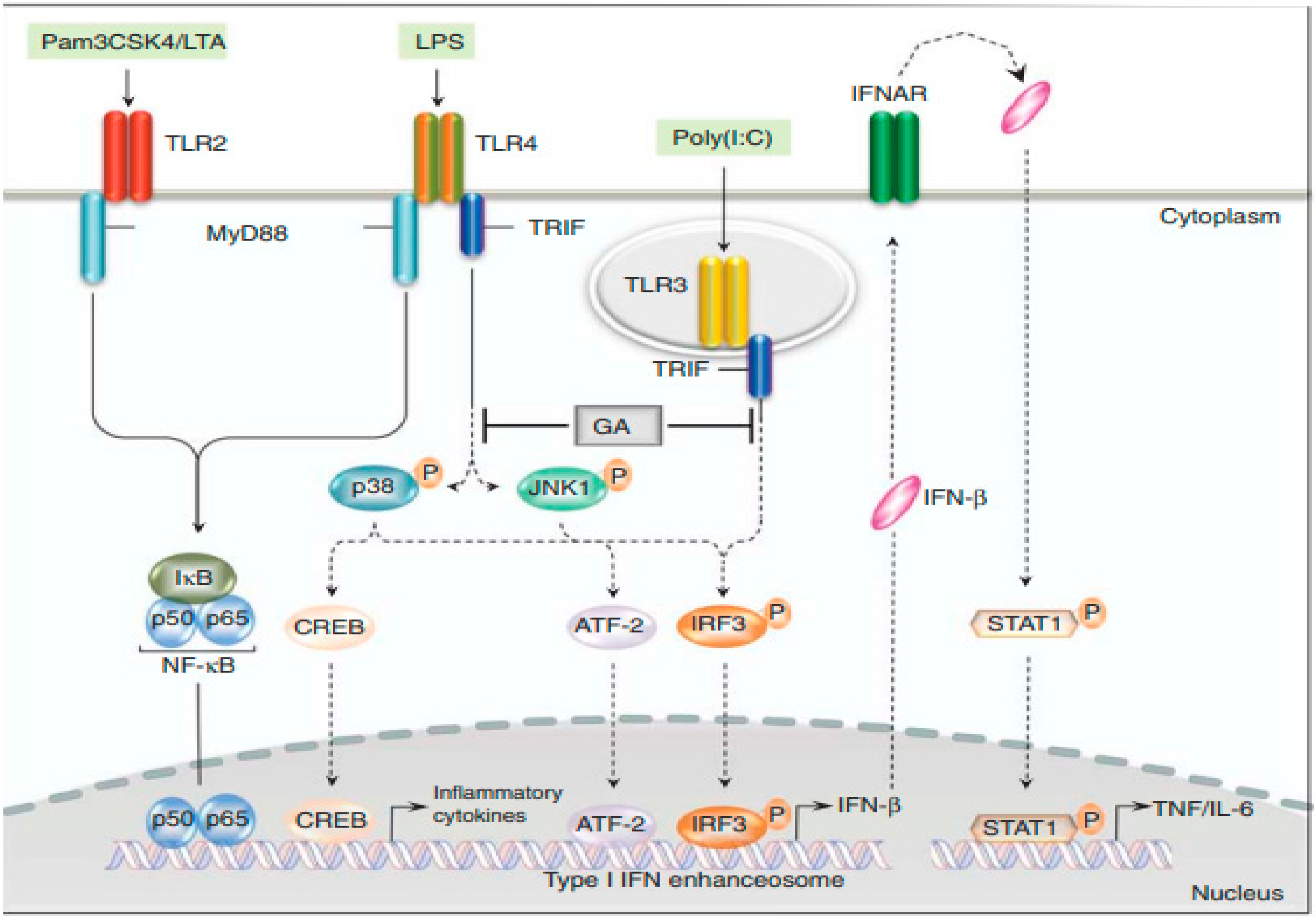

Figure 2. Glatiramer Acetate Modulates Type I Interferon production [50].
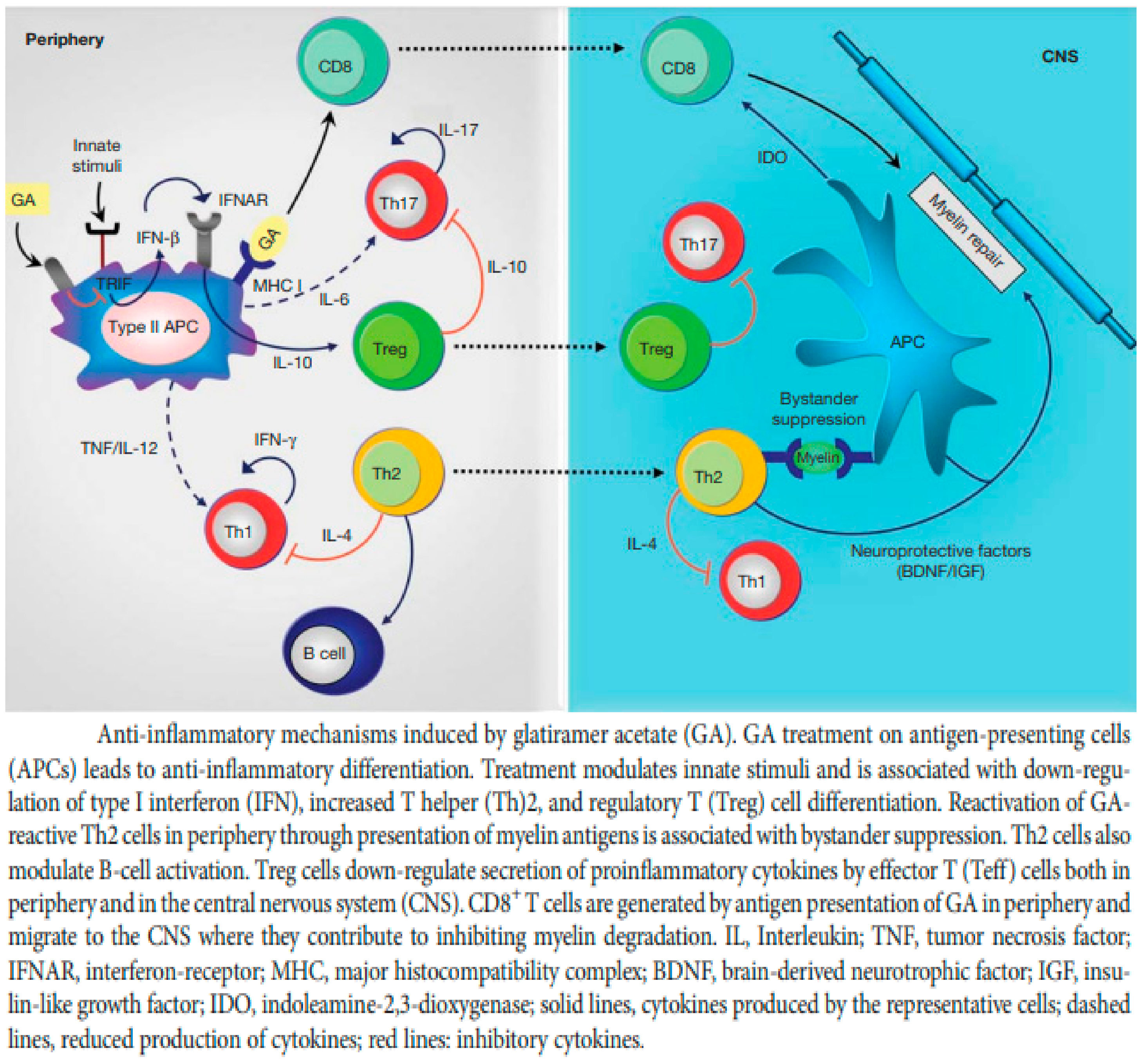
Figure 3. Anti-inflammatory Mechanisms Induced by Glatiramer Acetate [52].
5.3. Treatment of Clinically Isolated Syndromes
In this entry, researchers are reviewing some of the trials that specifically addressed clinically isolated syndromes such as optic neuritis. These are mostly older trials. Researchers will not cover all multiple sclerosis treatments in detail, but acknowledge that several newer DMTs for MS have class I evidence for MS, and have approval for treatment of both MS and CIS. This evidence (and the FDA approval of these medications) is based on MS trials, not specifically CIS/optic neuritis trials, and will not be reviewed in detail. Thus, in clinical practice, a number of additional MS medicines may be used for high-risk CIS patients, likely with good efficacy, although they were not specifically investigated in the CIS situation. It is beyond the scope of this entry to discuss all such treatment options.
The goal of MS treatment is to delay the onset of additional clinical relapses and possibly long-term disability. The first opportunity to initiate disease-modifying therapy in patients with MS may actually be when they are in the clinically isolated syndrome (CIS) stage, i.e., before conversion to clinically definite MS (CDMS). Since 1993, when interferon beta-1b was approved for MS, a growing number of disease-modifying therapies (DMTs) have become available. The goal of DMTs is to decrease the frequency of clinical relapses, lessen the number of new and active multiple sclerosis lesions on MRI, and, in the long term, to slow the progression of neurologic impairment. Since the approval of natalizumab as the first highly active DMT, the ultimate goal of “no evidence of disease activity” (NEDA) has become attainable for many patients. While the treatment of MS is beyond the scope of this entry, the evidence for initiating MS DMTs after CIS is discussed.
Most DMTs approved for MS are also approved for the treatment of CIS. However, only a few DMTs have specifically been evaluated in clinical trials to treat CIS (including ON) and to delay the onset of clinically definite MS, including interferons and glatiramer acetate [53][54][55][56][57][58]. In all trials, the patients who received the active drug developed a second neurologic manifestation (definite multiple sclerosis) less frequently, and (if at all) at a later time, than those given the placebo. Even after a second episode, treated patients had a significantly lower annual rate of relapse for the duration of the follow-up period. Neurologic impairment was generally relatively mild and not significantly different between the two groups.
Interferons and glatiramer acetate have been approved for the treatment of CIS, including ON with two or more inactive typical lesions of multiple sclerosis on MRI. CHAMPS (Controlled High-Risk Subjects Avonex Multiple Sclerosis Prevention Study) was a randomized, double-blind trial involving 383 patients with an initial, acute monosymptomatic demyelinating event—unilateral ON, incomplete transverse myelitis (TM), or brainstem/cerebellar—and at least 2 silent T2 lesions on brain MRI [59]. The patients were randomized to weekly intramuscular interferon β-1a (IFN-b1a) or a placebo. The treatment group experienced a 44% reduction in the rate of development of CDMS compared with the placebo group over 3 years of follow-ups. There were statistically significant beneficial effects on all MRI parameters for the treatment group, including a decrease in T2 lesion development, gadolinium-enhancing lesions, and T2 lesion volume. The 10-year follow-up showed that patients treated immediately after their first episode had a significantly lesser chance of experiencing a second attack compared to those who had delayed treatment. Based on these results, FDA extended its approval of intramuscular IFN-b1a to include patients with CIS deemed to be at high risk for MS. The most common side effects associated with interferons are flu-like symptoms, including myalgia, fever, fatigue, headache, chills, nausea, vomiting, pain, and asthenia [59].
The PRISM (Prevention of Relapses and Disability by Interferon β-1a Subcutaneously in Multiple Sclerosis) trial assessed the efficacy of interferon (IFN)-β1a compared to the placebo, in dosages of 22 μg and 44 μg given subcutaneously in relapsing-remitting MS patients; both treatment groups had fewer relapses [60]. The Early Treatment of Multiple Sclerosis (ETOMS) trial showed that weekly subcutaneous IFN-β1a reduced the conversion to CDMS over 2 years to 34% vs. 45% for the placebo; a post hoc analysis found that the treatment group had a reduced rate of brain atrophy compared with those on the placebo [61]. The BENEFIT (Betaseron in Newly Emerging Multiple Sclerosis for Initial Treatment) study included patients with a single neurologic event and at least 2 clinically silent MRI lesions; in a 24-month study period, the standard dose of IFN-β1 was seen to reduce the risk of MS by 50%. Furthermore, open-label extension studies from the original CHAMPS and BENEFIT cohorts have suggested a possible long-term benefit from the early initiation of disease modifying treatments [62]. The CHAMPIONS (Controlled High-Risk Avonex Multiple Sclerosis Prevention Study in Ongoing Neurologic Surveillance) trial concluded that a delay in treatment by up to 3 years after a first clinical demyelinating attack could lead to an earlier time for CDMS but did not show a long-term effect on the development of new MRI T2-weighted lesions or long-term disability [63]. The REFLEX (REbif FLEXible dosing in early MS) trial evaluated 517 patients with CIS and at least two clinically silent T2 lesions on brain MRI. At two years, the probability of MS diagnosed by the McDonald criteria was significantly lower with subcutaneous interferon β-1a 44 mcg dosed either three times a week or once a week (63 and 76 percent, vs. 86 percent for the placebo). In the subsequent extension phase of the trial, all patients (n = 403) received interferon β-1a. At five years, the group assigned to interferon β-1a treatment in the placebo-controlled phase (i.e., early treatment) continued to have a reduced probability of conversion to MS and fewer new MRI lesions compared with the group whose treatment was delayed for up to two years [64][65][66].
Glatiramer acetate is an immunomodulator used to reduce relapse frequency in relapsing–remitting multiple sclerosis [39]. The PreCISe (Early GA Treatment in Delaying Conversion to CDMS in Participants Presenting with a Clinically Isolated Syndrome) trial showed a reduced conversion to CDMS (25%) in patients treated with 20 mg of glatiramer acetate subcutaneously daily compared to 43% for the placebo [63].
Teriflunomide also reduces the risk of progression to multiple sclerosis, as has been shown in the TOPIC (Teriflunomide Vs. Placebo in Patients With First Clinical Symptom of Multiple Sclerosis) trial, where 618 adults with a CIS were randomly assigned in a 1:1:1 ratio for treatment with 14 mg of oral teriflunomide daily, 7 mg of teriflunomide daily, or the placebo for up to 108 weeks, with a median treatment duration of over 70 weeks. The agent reduced the risk of relapse-defining CDMS at both the 14 mg dose and the 7 mg dose. The exact mechanisms by which teriflunomide works in MS are not established; it is an oral dihydroorotate dehydrogenase inhibitor that interferes with de novo synthesis of pyrimidines and thus inhibits the proliferation of rapidly dividing cells such as autoreactive T and B cells [64]. The most common adverse effects of teriflunomide were elevated alanine aminotransferase (ALT) levels, diarrhea, hair thinning, paresthesia, and upper respiratory tract infections. Teriflunomide is associated with increased risk for hepatotoxicity and teratogenicity and should not be given to patients with liver disease or women who are pregnant. Full immunization coverage is required prior to treatment initiation [53][54][55][67]. In addition, intravenous immune globulin and minocycline have been studied for the treatment of CIS or the first demyelinating event, but are not established as effective [56][57][58].
The early treatment of CIS is not favored by all experts. The decision whether to initiate treatment for CIS has to consider that not all patients go on to develop any additional relapses or lesions, and that the evidence base showing that the early treatment of CIS will prevent long-term disability is very limited. Patients should be informed of the potential benefits, risks, and uncertainties, and participate in decision making [62]. However, once a diagnosis of CDMS is made, the early initiation of treatment is recommended.
References
- Menon, V.; Saxena, R.; Misra, R.; Phuljhele, S. Management of optic neuritis. Indian J. Ophthalmol. 2011, 59, 117–122.
- Morrow, S.A.; Fraser, J.A.; Day, C.; Bowman, D.; Rosehart, H.; Kremenchutzky, M.; Nicolle, M. Effect of treating acute optic neuritis with bioequivalent oral vs intravenous corticosteroids—A randomized clinical trial. JAMA Neurol. 2018, 75, 690–696.
- Huang, W.J.; Chen, W.W.; Zhang, X. Multiple sclerosis: Pathology, diagnosis and treatments. Exp. Ther. Med. 2017, 13, 3163–3166.
- Compston, A.; Coles, A. Multiple sclerosis. Lancet 2008, 372, 1502–1517.
- Wingerchuk, D.; Lucchinetti, C.; Noseworthy, J. Multiple Sclerosis: Current Pathophysiological Concepts. Lab. Investig. 2001, 81, 263–281.
- Lucchinetti, C.; Brück, W.; Parisi, J.; Scheithauer, B.; Rodriguez, M.; Lassmann, H. Heterogeneity of multiple sclerosis lesions: Implications for the pathogenesis of demyelination. Ann. Neurol. 2000, 47, 707–717.
- Traugott, U.; Reinherz, E.L.; Raine, C.S. Multiple sclerosis. Distribution of T cells, T cell subsets and Ia-positive macrophages in lesions of different ages. J. Neuroimmunol. 1983, 4, 201–221.
- Ferguson, B.; Matyszak, M.K.; Esiri, M.M.; Perry, V.H. Axonal damage in acute multiple sclerosis lesions. Brain 1997, 120, 393–399.
- Bitsch, A.; Schuchardt, J.; Bunkowski, S.; Kuhlmann, T.; Brück, W. Acute axonal injury in multiple sclerosis. Correlation with demyelination and inflammation. Brain 2000, 123, 1174–1183.
- Hu, D.; Ikizawa, K.; Lu, L.; Sanchirico, M.E.; Shinohara, M.L.; Cantor, H. Analysis of regulatory CD8 T cells in Qa-1-deficient mice. Nat. Immunol. 2004, 5, 516–523.
- Toosy, A.T.; Mason, D.F.; Miller, D.H. Optic neuritis. Lancet Neurol. 2014, 13, 83–99.
- Babbe, H.; Roers, A.; Waisman, A.; Lassmann, H.; Goebels, N.; Hohlfeld, R.; Friese, M.; Schröder, R.; Deckert, M.; Schmidt, S.; et al. Clonal expansions of CD8(+) T cells dominate the T cell infiltrate in active multiple sclerosis lesions as shown by micromanipulation and single cell polymerase chain reaction. J. Exp. Med. 2000, 192, 393–404.
- Carlström, K.E.; Zhu, K.; Ewing, E.; Krabbendam, I.E.; Harris, R.A.; Falcão, A.M.; Jagodic, M.; Castelo-Branco, G.; Piehl, F. Gsta4 controls apoptosis of differentiating adult oligodendrocytes during homeostasis and remyelination via the mitochondria-associated Fas-Casp8-Bid-axis. Nat. Commun. 2020, 11, 4071.
- Chamberlain, K.A.; Chapey, K.S.; Nanescu, S.E.; Huang, J.K. Creatine enhances mitochondrial-mediated oligodendrocyte survival after demyelinating injury. J. Neurosci. 2017, 37, 1479–1492.
- Jin, J.; Smith, M.D.; Kersbergen, C.J.; Kam, T.-I.; Viswanathan, M.; Martin, K.; Dawson, T.M.; Dawson, V.L.; Zack, D.J.; Whartenby, K.; et al. Glial pathology and retinal neurotoxicity in the anterior visual pathway in experimental autoimmune encephalomyelitis. Acta Neuropathol. Commun. 2019, 7, 125.
- Cannella, B.; Raine, C.S. The adhesion molecule and cytokine profile of multiple sclerosis lesions. Ann. Neurol. 1995, 37, 424–435.
- Kuhlmann, T.; Miron, V.; Cui, Q.; Wegner, C.; Antel, J.; Brück, W. Differentiation block of oligodendroglial progenitor cells as a cause for remyelination failure in chronic multiple sclerosis. Brain 2008, 131 Pt 7, 1749–1758.
- Goodkin, D.E. The Natural History of Multiple Sclerosis. In Treatment of Multiple Sclerosis; Clinical Medicine and the Nervous System; Rudick, R.A., Goodkin, D.E., Eds.; Springer: London, UK, 1992.
- Koopmans, R.A.; Li, D.K.B.; Oger, J.J.F.; Kastrukoff, L.F.; Jardine, C.; Costley, L.; Hall, S.; Grochowski, E.W.; Paty, D.W. Chronic progressive multiple sclerosis: Serial magnetic resonance brain imaging over six months. Ann. Neurol. 1989, 26, 248–256.
- Prineas, J.W.; Barnard, R.O.; Kwon, E.E.; Sharer, L.R.; Cho, E.S. Multiple sclerosis: Remyelination of nascent lesions. Ann. Neurol. 1993, 33, 137–151.
- Lotan, I.; Hellmann, M.A.; Benninger, F.; Stiebel-Kalish, H.; Steiner, I. Recurrent optic neuritis—Different patterns in multiple sclerosis, neuromyelitis optica spectrum disorders and MOG-antibody disease. J. Neuroimmunol. 2018, 324, 115–118.
- Burman, J.; Raininko, R.; Fagius, J. Bilateral and recurrent optic neuritis in multiple sclerosis. Acta Neurol. Scand. 2011, 123, 207–210.
- Quintana, F.J.; Patel, B.; Yeste, A.; Nyirenda, M.; Kenison, J.; Rahbari, R.; Fetco, D.; Hussain, M.; O’Mahony, J.; Magalhaes, S.; et al. Canadian Pediatric Demyelinating Disease Network. Epitope spreading as an early pathogenic event in pediatric multiple sclerosis. Neurology 2014, 83, 2219–2226.
- Frohman, E.M.; Racke, M.K.; Raine, C.S. Multiple sclerosis—The plaque and its pathogenesis. N. Engl. J. Med. 2006, 354, 942–955.
- Qin, Y.; Duquette, P.; Zhang, Y.; Talbot, P.; Poole, R.; Antel, J. Clonal expansion and somatic hypermutation of V (H) genes of B cells from cerebrospinal fluid in multiple sclerosis. J. Clin. Investig. 1998, 102, 1045–1050.
- Noseworthy, J.H.; Lucchinetti, C.; Rodriguez, M.; Weinshenker, B.G. Multiple sclerosis. N. Engl. J. Med. 2000, 343, 938–952.
- Sriram, S.; Steiner, I. Experimental allergic encephalomyelitis: A misleading model of multiple sclerosis. Ann. Neurol. 2005, 58, 939–945.
- Hellmann, M.A.; Steiner, I.; Mosberg-Galili, R. Sudden sensorineural hearing loss in multiple sclerosis: Clinical course and possible pathogenesis. Acta Neurol. Scand. 2011, 124, 245–249.
- Lemus, H.N.; Warrington, A.E.; Rodriguez, M. Multiple Sclerosis: Mechanisms of Disease and Strategies for Myelin and Axonal Repair. Neurol. Clin. 2018, 36, 1–11.
- Nancy, J.; Newman, M.D. Atlanta, Georgia, the Optic Neuritis Treatment Trial. Commentary, AAO. Available online: https://www.aaojournal.org/article/S0161-6420(19)32364-4/pdf (accessed on 2 February 2022).
- Beck, R.W.; Gal, R.L. Treatment of acute optic neuritis: A summary of findings from the optic neuritis treatment trial. Arch. Ophthalmol. 2008, 126, 994–995.
- Beck, R.W.; Cleary, P.A.; Anderson, M.M., Jr.; Keltner, J.L.; Shults, W.T.; Kaufman, D.I.; Buckley, E.G.; Corbett, J.J.; Kupersmith, M.J.; Miller, N.R.; et al. A randomized controlled trail of corticosteroids in the treatment of acute optic neuritis. N. Engl. J. Med. 1992, 326, 581–588.
- Optic Neuritis Study Group. Visual function 5 years after optic neuritis: Experience of the Optic Neuritis Treatment Trial. Arch. Ophthalmol. 1997, 115, 1545–1552.
- Gal, R.L.; Vedula, S.S.; Beck, R. Corticosteroids for treating optic neuritis. Cochrane Database Syst. Rev. 2015, 2015, CD001430.
- Petzold, A.; Braithwaite, T.; van Oosten, B.W.; Balk, L.; Martinez-Lapiscina, E.H.; Wheeler, R.; Wiegerinck, N.; Waters, C.; Plant, G.T. Case for a new corticosteroid treatment trial in optic neuritis: Review of updated evidence. J. Neurol. Neurosurg. Psychiatry 2020, 91, 9–14.
- Stiebel-Kalish, H.; Hellmann, M.A.; Mimouni, M.; Paul, F.; Bialer, O.; Bach, M.; Lotan, I. Does time equal vision in the acute treatment of a cohort of AQP4 and MOG optic neuritis? Neurol. Neuroimmunol. Neuroinflamm. 2019, 6, e572.
- Bsteh, G.; Berek, K.; Hegen, H.; Teuchner, B.; Buchmann, A.; Voortman, M.M.; Auer, M.; Zinganell, A.; Di Pauli, F.; Deisenhammer, F.; et al. Serum neurofilament levels correlate with retinal nerve fiber layer thinning in multiple sclerosis. Mult. Scler. J. 2019, 26, 1682–1690.
- Osinga, E.; van Oosten, B.; de Vries-Knoppert, W.; Petzold, A. Time is vision in recurrent optic neuritis. Brain Res. 2017, 1673, 95–101.
- Phuljhele, S.; Kedar, S.; Saxena, R. Approach to optic neuritis: An update. Indian J. Ophthalmol. 2021, 69, 2266–2276.
- Horton, L.; Bennett, J.L. Acute Management of Optic Neuritis: An Evolving Paradigm. J. Neuroophthalmol. 2018, 38, 358–367.
- Wilhelm, H.; Schabet, M. Continuing medical education the diagnosis and treatment of optic neuritis. Dtsch. Arztebl. Int. 2015, 112, 616–626.
- Le Page, E.; Veillard, D.; Laplaud, D.A.; Hamonic, S.; Wardi, R.; Lebrun, C.; Zagnoli, F.; Wiertlewski, S.; Deburghgraeve, V.; Coustans, M.; et al. Oral versus intravenous high-dose methylprednisolone for treatment of relapses inpatients with multiple sclerosis (COPOUSEP): A randomised, controlled, double-blind, non-inferiority trial. Lancet 2015, 386, 974–981.
- Bonnan, M.; Cabre, P. Plasma Exchange in Severe Attacks of Neuromyelitis Optica. Mult. Scler. Int. 2012, 2012, 787630.
- Bennett, J.L. Optic Neuritis. Continuum 2019, 25, 1236–1264.
- Jakimovski, D.; Kolb, C.; Ramanathan, M.; Zivadinov, R.; Weinstock-Guttman, B. Interferon β for Multiple Sclerosis. Cold Spring Harb. Perspect. Med. 2018, 8, a032003.
- Dhib-Jalbut, S.; Marks, S. Interferon-beta mechanisms of action in multiple sclerosis. Neurology 2010, 74 (Suppl. S1), S17–S24.
- Rudick, R.A.; Ransohoff, R.M.; Lee, J.C.; Peppler, R.; Yu, M.; Mathisen, P.M.; Tuohy, V.K. In vivo effects of interferon beta-1a on immunosuppressive cytokines in multiple sclerosis. Neurology 1998, 50, 1294–1300.
- Hartrich, L.; Weinstock-Guttman, B.; Hall, D.; Badgett, D.; Baier, M.; Patrick, K.; Feichter, J.; Hong, J.; Ramanathan, M. Dynamics of immune cell trafficking in interferon-β treated multiple sclerosis patients. J. Neuroimmunol. 2003, 139, 84–92.
- Rizzo, F.; Giacomini, E.; Mechelli, R.; Buscarinu, M.C.; Salvetti, M.; Severa, M.; Coccia, E.M. Interferon-β therapy specifically reduces pathogenic memory B cells in multiple sclerosis patients by inducing a FAS-mediated apoptosis. Immunol. Cell. Biol. 2016, 94, 886–894.
- Molnarfi, N.; Prod’homme, T.; Schulze-Topphoff, U.; Spencer, C.M.; Weber, M.S.; Patarroyo, J.C.; Lalive, P.H.; Zamvil, S.S. Glatiramer acetate treatment negatively regulates type I interferon signaling. Neurol. Neuroimmunol. Neuroinflamm. 2015, 2, e179.
- Weber, M.S.; Hohlfeld, R.; Zamvil, S.S. Mechanism of action of glatiramer acetate in treatment of multiple sclerosis. Neurotherapeutics 2007, 4, 647–653.
- Prod’homme, T.; Zamvil, S.S. The Evolving Mechanisms of Action of Glatiramer Acetate. Cold Spring Harb. Perspect. Med. 2019, 9, a029249.
- Polman, C.H.; Reingold, S.C.; Banwell, B.; Clanet, M.; Cohen, J.A.; Filippi, M.; Fujihara, K.; Havrdova, E.; Hutchinson, M.; Kappos, L.; et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann. Neurol. 2011, 69, 292–302.
- Kieseier, B.C.; Benamor, M. Pregnancy outcomes following maternal and paternal exposure to teriflunomide during treatment for relapsing-remitting multiple sclerosis. Neurol. Ther. 2014, 3, 133–138.
- Andersen, J.B.; Moberg, J.Y.; Spelman, T.; Magyari, M. Pregnancy Outcomes in Men and Women Treated with Teriflunomide. A Population-Based Nationwide Danish Register Study. Front. Immunol. 2018, 9, 2706.
- Fazekas, F.; Lublin, F.D.; Li, D.; Freedman, M.S.; Hartung, H.P.; Rieckmann, P.; Sørensen, P.S.; Maas-Enriquez, M.; Sommerauer, B.; Hanna, K.; et al. Intravenous immunoglobulin in relapsing-remitting multiple sclerosis: A dose-finding trial. Neurology 2008, 71, 265–271.
- Sørensen, P.S.; Sellebjerg, F.; Lycke, J.; Färkkilä, M.; Créange, A.; Lund, C.G.; Schluep, M.; Frederiksen, J.L.; Stenager, E.; Pfleger, C.; et al. Minocycline added to subcutaneous interferon β-1a in multiple sclerosis: Randomized Recycline study. Eur. J. Neurol. 2016, 23, 861–870.
- Metz, L.M.; Li, D.K.B.; Traboulsee, A.L.; Duquette, P.; Eliasziw, M.; Cerchiaro, G.; Greenfield, J.; Riddehough, A.; Yeung, M.; Kremenchutzky, M.; et al. Trial of Minocycline in a Clinically Isolated Syndrome of Multiple Sclerosis. N. Engl. J. Med. 2017, 376, 2122–2133.
- Jacobs, L.D.; Beck, R.W.; Simon, J.H.; Kinkel, R.P.; Brownscheidle, C.M.; Murray, T.J.; Simonian, N.A.; Slasor, P.J.; Sandrock, A.W.; The CHAMPS Study Group. Intramuscular interferon beta-1a therapy initiated during a first demyelinating event in multiple sclerosis. N. Engl. J. Med. 2000, 343, 898–904.
- Goodin, D.S.; Bates, D. Treatment of early multiple sclerosis: The value of treatment initiation after a first clinical episode. Mult. Scler. 2009, 15, 1175–1182.
- Comi, G.; Filippi, M.; Barkhof, F.; Durelli, L.; Edan, G.; Fernández, O.; Hartung, H.; Seeldrayers, P.; Sørensen, P.S.; Rovaris, M.; et al. Effect of early interferon treatment on conversion to definite multiple sclerosis: A randomised study. Lancet 2001, 357, 1576–1582.
- Efendi, H. Clinically Isolated Syndromes: Clinical Characteristics, Differential Diagnosis, and Management. Noro Psikiyatr. Ars. 2015, 52 (Suppl. S1), S1–S11.
- Marcus, J.F.; Waubant, E.L. Updates on Clinically Isolated Syndrome and Diagnostic Criteria for Multiple Sclerosis. Neurohospitalist 2013, 3, 65–80.
- Gold, R.; Wolinsky, J.S. Pathophysiology of multiple sclerosis and the place of teriflunomide. Acta Neurol. Scand. 2011, 124, 75–84.
- Comi, G.; De Stefano, N.; Freedman, M.S.; Barkhof, F.; Polman, C.H.; Uitdehaag, B.M.J.; Casset-Semanaz, F.; Hennessy, B.; Moraga, M.S.; Rocak, S.; et al. Comparison of two dosing frequencies of subcutaneous interferon beta-1a in patients with a first clinical demyelinating event suggestive of multiple sclerosis (REFLEX): A phase 3 randomised controlled trial. Lancet Neurol. 2012, 11, 33.
- Comi, G.; De Stefano, N.; Freedman, M.S.; Barkhof, F.; Uitdehaag, B.M.; de Vos, M.; Marhardt, K.; Chen, L.; Issard, D.; Kappos, L. Subcutaneous interferon β-1a in the treatment of clinically isolated syndromes: 3-year and 5-year results of the phase III dosing frequency-blind multicentre REFLEXION study. J. Neurol. Neurosurg. Psychiatry 2017, 88, 285–294.
- Miller, A.E.; Wolinsky, J.S.; Kappos, L.; Comi, G.; Freedman, M.S.; Olsson, T.P.; Bauer, D.; Benamor, M.; Truffinet, P.; O’Connor, P.W.; et al. Oral teriflunomide for patients with a first clinical episode suggestive of multiple sclerosis (TOPIC): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Neurol. 2014, 13, 977–986.
More
Information
Subjects:
Ophthalmology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
923
Revisions:
2 times
(View History)
Update Date:
20 Sep 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No