Optic neuritis (ON) is an inflammatory condition involving the optic nerve. Several important typical and atypical ON variants are now recognized. Typical ON has a more favorable prognosis; it can be idiopathic or represent an early manifestation of demyelinating diseases, mostly multiple sclerosis (MS). The atypical spectrum includes entities such as antibody-driven ON associated with neuromyelitis optica spectrum disorder (NMOSD) and myelin oligodendrocyte glycoprotein antibody disease (MOGAD), chronic/relapsing inflammatory optic neuropathy (CRION), and sarcoidosis-associated ON. Appropriate and timely diagnosis is essential to rapidly decide on the appropriate treatment, maximize visual recovery, and minimize recurrences.
- atypical optic neuritis treatment
- typical optic neuritis
- MOG
- NMO
- MS prevention
1. Introduction
2. Typical Optic Neuritis
3. Pathophysiology of ON
4. Acute Treatment of Typical Optic Neuritis/Clinically Isolated Syndrome
5. Long-Term Treatment: Immune Prophylaxis against Optic Neuritis Relapses/Progression to Multiple Sclerosis
5.1. Mechanisms of Action in Interferon β in MS and Optic Neuritis
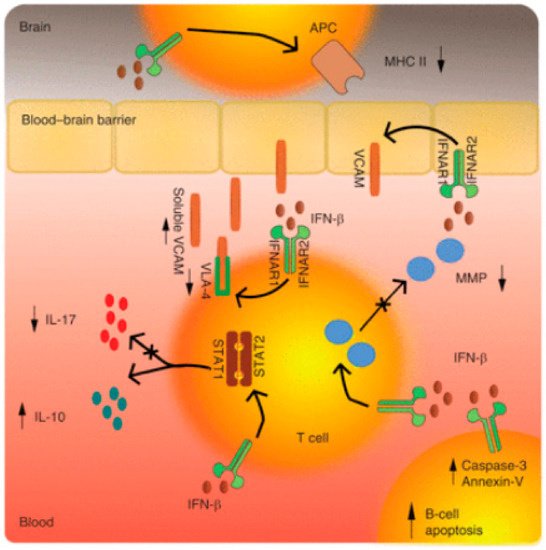

5.2. Glatiramer Acetate (GA)
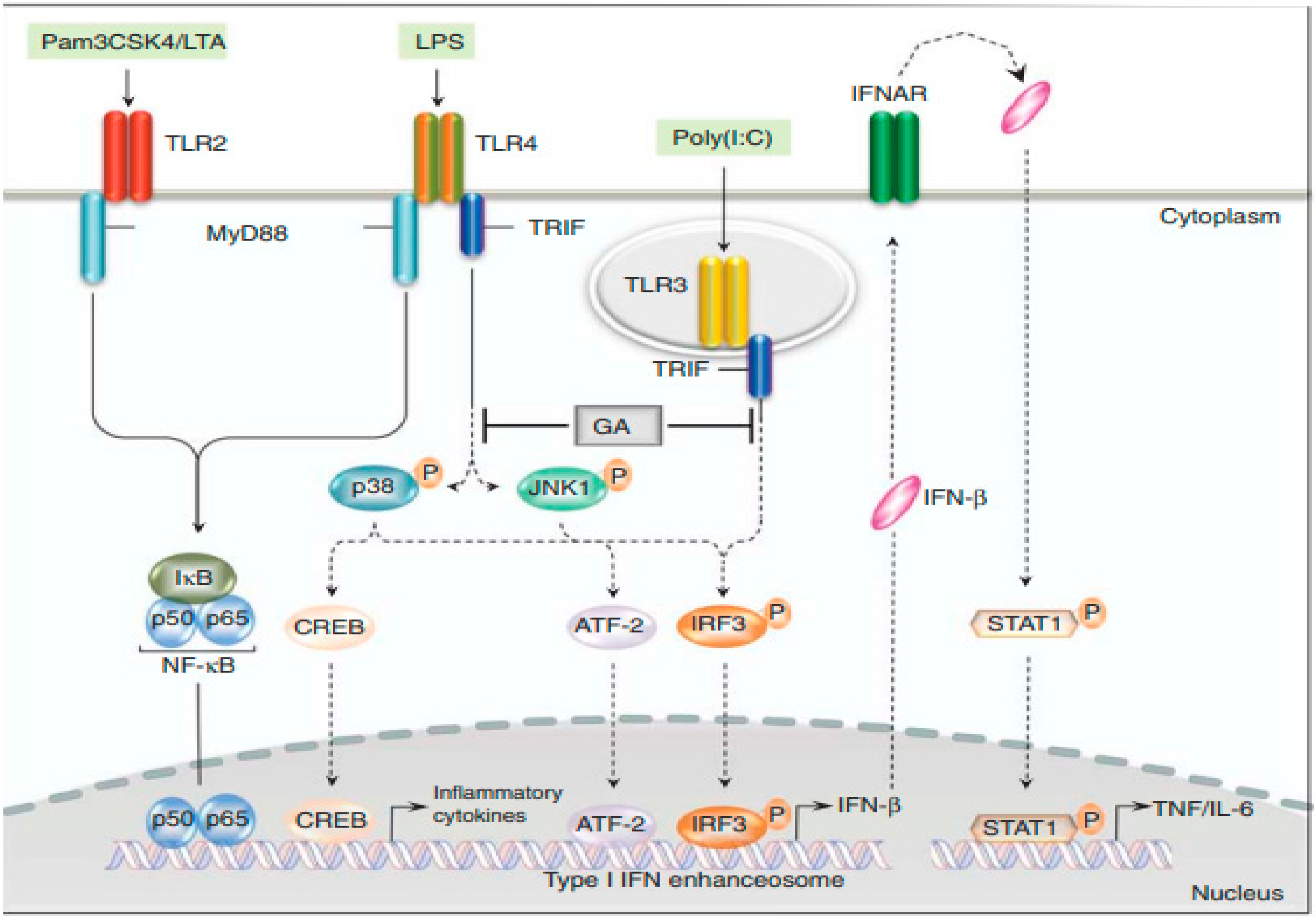

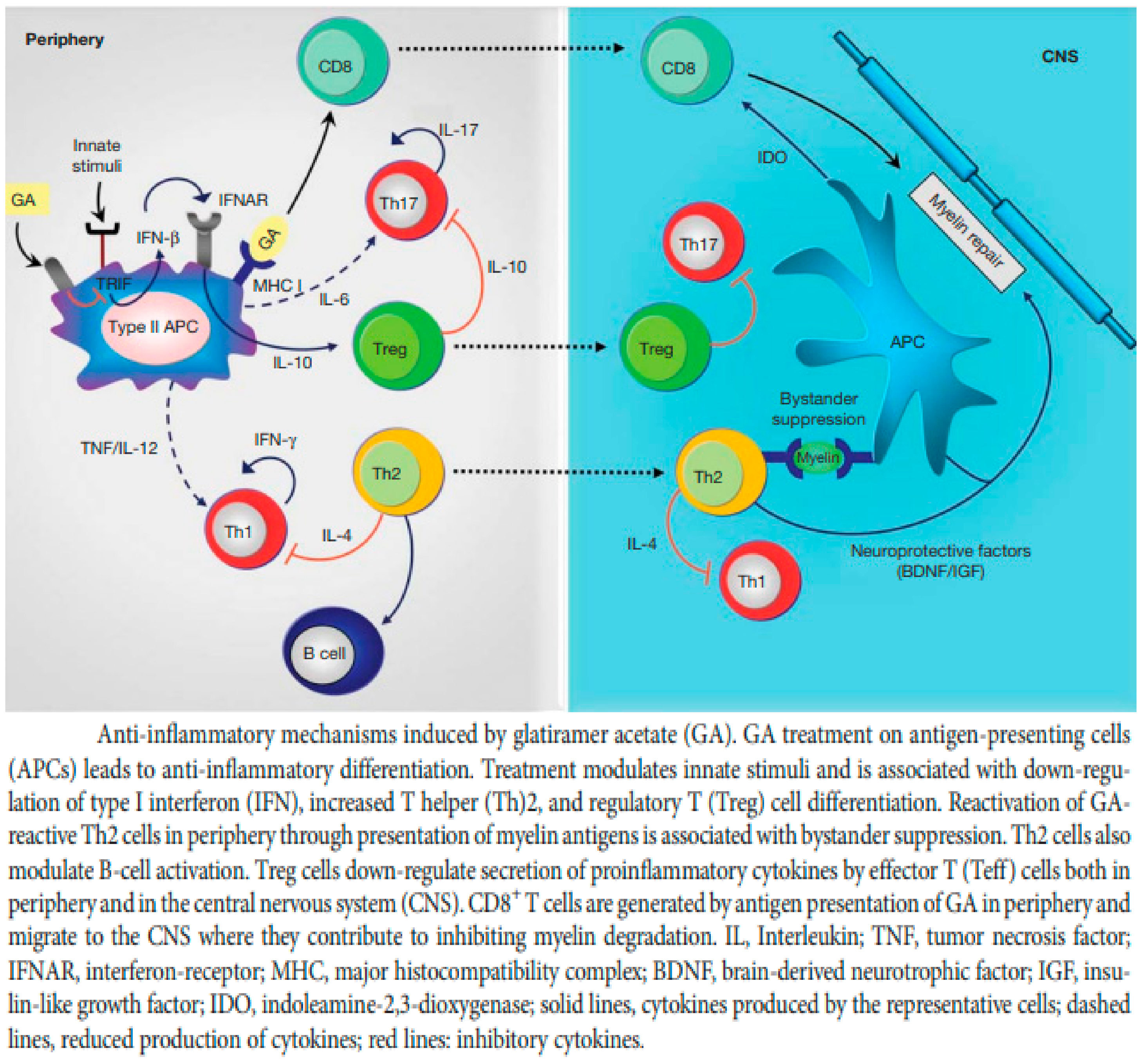
5.3. Treatment of Clinically Isolated Syndromes
This entry is adapted from the peer-reviewed paper 10.3390/ijms23179769
References
- Menon, V.; Saxena, R.; Misra, R.; Phuljhele, S. Management of optic neuritis. Indian J. Ophthalmol. 2011, 59, 117–122.
- Morrow, S.A.; Fraser, J.A.; Day, C.; Bowman, D.; Rosehart, H.; Kremenchutzky, M.; Nicolle, M. Effect of treating acute optic neuritis with bioequivalent oral vs intravenous corticosteroids—A randomized clinical trial. JAMA Neurol. 2018, 75, 690–696.
- Huang, W.J.; Chen, W.W.; Zhang, X. Multiple sclerosis: Pathology, diagnosis and treatments. Exp. Ther. Med. 2017, 13, 3163–3166.
- Compston, A.; Coles, A. Multiple sclerosis. Lancet 2008, 372, 1502–1517.
- Wingerchuk, D.; Lucchinetti, C.; Noseworthy, J. Multiple Sclerosis: Current Pathophysiological Concepts. Lab. Investig. 2001, 81, 263–281.
- Lucchinetti, C.; Brück, W.; Parisi, J.; Scheithauer, B.; Rodriguez, M.; Lassmann, H. Heterogeneity of multiple sclerosis lesions: Implications for the pathogenesis of demyelination. Ann. Neurol. 2000, 47, 707–717.
- Traugott, U.; Reinherz, E.L.; Raine, C.S. Multiple sclerosis. Distribution of T cells, T cell subsets and Ia-positive macrophages in lesions of different ages. J. Neuroimmunol. 1983, 4, 201–221.
- Ferguson, B.; Matyszak, M.K.; Esiri, M.M.; Perry, V.H. Axonal damage in acute multiple sclerosis lesions. Brain 1997, 120, 393–399.
- Bitsch, A.; Schuchardt, J.; Bunkowski, S.; Kuhlmann, T.; Brück, W. Acute axonal injury in multiple sclerosis. Correlation with demyelination and inflammation. Brain 2000, 123, 1174–1183.
- Hu, D.; Ikizawa, K.; Lu, L.; Sanchirico, M.E.; Shinohara, M.L.; Cantor, H. Analysis of regulatory CD8 T cells in Qa-1-deficient mice. Nat. Immunol. 2004, 5, 516–523.
- Toosy, A.T.; Mason, D.F.; Miller, D.H. Optic neuritis. Lancet Neurol. 2014, 13, 83–99.
- Babbe, H.; Roers, A.; Waisman, A.; Lassmann, H.; Goebels, N.; Hohlfeld, R.; Friese, M.; Schröder, R.; Deckert, M.; Schmidt, S.; et al. Clonal expansions of CD8(+) T cells dominate the T cell infiltrate in active multiple sclerosis lesions as shown by micromanipulation and single cell polymerase chain reaction. J. Exp. Med. 2000, 192, 393–404.
- Carlström, K.E.; Zhu, K.; Ewing, E.; Krabbendam, I.E.; Harris, R.A.; Falcão, A.M.; Jagodic, M.; Castelo-Branco, G.; Piehl, F. Gsta4 controls apoptosis of differentiating adult oligodendrocytes during homeostasis and remyelination via the mitochondria-associated Fas-Casp8-Bid-axis. Nat. Commun. 2020, 11, 4071.
- Chamberlain, K.A.; Chapey, K.S.; Nanescu, S.E.; Huang, J.K. Creatine enhances mitochondrial-mediated oligodendrocyte survival after demyelinating injury. J. Neurosci. 2017, 37, 1479–1492.
- Jin, J.; Smith, M.D.; Kersbergen, C.J.; Kam, T.-I.; Viswanathan, M.; Martin, K.; Dawson, T.M.; Dawson, V.L.; Zack, D.J.; Whartenby, K.; et al. Glial pathology and retinal neurotoxicity in the anterior visual pathway in experimental autoimmune encephalomyelitis. Acta Neuropathol. Commun. 2019, 7, 125.
- Cannella, B.; Raine, C.S. The adhesion molecule and cytokine profile of multiple sclerosis lesions. Ann. Neurol. 1995, 37, 424–435.
- Kuhlmann, T.; Miron, V.; Cui, Q.; Wegner, C.; Antel, J.; Brück, W. Differentiation block of oligodendroglial progenitor cells as a cause for remyelination failure in chronic multiple sclerosis. Brain 2008, 131 Pt 7, 1749–1758.
- Goodkin, D.E. The Natural History of Multiple Sclerosis. In Treatment of Multiple Sclerosis; Clinical Medicine and the Nervous System; Rudick, R.A., Goodkin, D.E., Eds.; Springer: London, UK, 1992.
- Koopmans, R.A.; Li, D.K.B.; Oger, J.J.F.; Kastrukoff, L.F.; Jardine, C.; Costley, L.; Hall, S.; Grochowski, E.W.; Paty, D.W. Chronic progressive multiple sclerosis: Serial magnetic resonance brain imaging over six months. Ann. Neurol. 1989, 26, 248–256.
- Prineas, J.W.; Barnard, R.O.; Kwon, E.E.; Sharer, L.R.; Cho, E.S. Multiple sclerosis: Remyelination of nascent lesions. Ann. Neurol. 1993, 33, 137–151.
- Lotan, I.; Hellmann, M.A.; Benninger, F.; Stiebel-Kalish, H.; Steiner, I. Recurrent optic neuritis—Different patterns in multiple sclerosis, neuromyelitis optica spectrum disorders and MOG-antibody disease. J. Neuroimmunol. 2018, 324, 115–118.
- Burman, J.; Raininko, R.; Fagius, J. Bilateral and recurrent optic neuritis in multiple sclerosis. Acta Neurol. Scand. 2011, 123, 207–210.
- Quintana, F.J.; Patel, B.; Yeste, A.; Nyirenda, M.; Kenison, J.; Rahbari, R.; Fetco, D.; Hussain, M.; O’Mahony, J.; Magalhaes, S.; et al. Canadian Pediatric Demyelinating Disease Network. Epitope spreading as an early pathogenic event in pediatric multiple sclerosis. Neurology 2014, 83, 2219–2226.
- Frohman, E.M.; Racke, M.K.; Raine, C.S. Multiple sclerosis—The plaque and its pathogenesis. N. Engl. J. Med. 2006, 354, 942–955.
- Qin, Y.; Duquette, P.; Zhang, Y.; Talbot, P.; Poole, R.; Antel, J. Clonal expansion and somatic hypermutation of V (H) genes of B cells from cerebrospinal fluid in multiple sclerosis. J. Clin. Investig. 1998, 102, 1045–1050.
- Noseworthy, J.H.; Lucchinetti, C.; Rodriguez, M.; Weinshenker, B.G. Multiple sclerosis. N. Engl. J. Med. 2000, 343, 938–952.
- Sriram, S.; Steiner, I. Experimental allergic encephalomyelitis: A misleading model of multiple sclerosis. Ann. Neurol. 2005, 58, 939–945.
- Hellmann, M.A.; Steiner, I.; Mosberg-Galili, R. Sudden sensorineural hearing loss in multiple sclerosis: Clinical course and possible pathogenesis. Acta Neurol. Scand. 2011, 124, 245–249.
- Lemus, H.N.; Warrington, A.E.; Rodriguez, M. Multiple Sclerosis: Mechanisms of Disease and Strategies for Myelin and Axonal Repair. Neurol. Clin. 2018, 36, 1–11.
- Nancy, J.; Newman, M.D. Atlanta, Georgia, the Optic Neuritis Treatment Trial. Commentary, AAO. Available online: https://www.aaojournal.org/article/S0161-6420(19)32364-4/pdf (accessed on 2 February 2022).
- Beck, R.W.; Gal, R.L. Treatment of acute optic neuritis: A summary of findings from the optic neuritis treatment trial. Arch. Ophthalmol. 2008, 126, 994–995.
- Beck, R.W.; Cleary, P.A.; Anderson, M.M., Jr.; Keltner, J.L.; Shults, W.T.; Kaufman, D.I.; Buckley, E.G.; Corbett, J.J.; Kupersmith, M.J.; Miller, N.R.; et al. A randomized controlled trail of corticosteroids in the treatment of acute optic neuritis. N. Engl. J. Med. 1992, 326, 581–588.
- Optic Neuritis Study Group. Visual function 5 years after optic neuritis: Experience of the Optic Neuritis Treatment Trial. Arch. Ophthalmol. 1997, 115, 1545–1552.
- Gal, R.L.; Vedula, S.S.; Beck, R. Corticosteroids for treating optic neuritis. Cochrane Database Syst. Rev. 2015, 2015, CD001430.
- Petzold, A.; Braithwaite, T.; van Oosten, B.W.; Balk, L.; Martinez-Lapiscina, E.H.; Wheeler, R.; Wiegerinck, N.; Waters, C.; Plant, G.T. Case for a new corticosteroid treatment trial in optic neuritis: Review of updated evidence. J. Neurol. Neurosurg. Psychiatry 2020, 91, 9–14.
- Stiebel-Kalish, H.; Hellmann, M.A.; Mimouni, M.; Paul, F.; Bialer, O.; Bach, M.; Lotan, I. Does time equal vision in the acute treatment of a cohort of AQP4 and MOG optic neuritis? Neurol. Neuroimmunol. Neuroinflamm. 2019, 6, e572.
- Bsteh, G.; Berek, K.; Hegen, H.; Teuchner, B.; Buchmann, A.; Voortman, M.M.; Auer, M.; Zinganell, A.; Di Pauli, F.; Deisenhammer, F.; et al. Serum neurofilament levels correlate with retinal nerve fiber layer thinning in multiple sclerosis. Mult. Scler. J. 2019, 26, 1682–1690.
- Osinga, E.; van Oosten, B.; de Vries-Knoppert, W.; Petzold, A. Time is vision in recurrent optic neuritis. Brain Res. 2017, 1673, 95–101.
- Phuljhele, S.; Kedar, S.; Saxena, R. Approach to optic neuritis: An update. Indian J. Ophthalmol. 2021, 69, 2266–2276.
- Horton, L.; Bennett, J.L. Acute Management of Optic Neuritis: An Evolving Paradigm. J. Neuroophthalmol. 2018, 38, 358–367.
- Wilhelm, H.; Schabet, M. Continuing medical education the diagnosis and treatment of optic neuritis. Dtsch. Arztebl. Int. 2015, 112, 616–626.
- Le Page, E.; Veillard, D.; Laplaud, D.A.; Hamonic, S.; Wardi, R.; Lebrun, C.; Zagnoli, F.; Wiertlewski, S.; Deburghgraeve, V.; Coustans, M.; et al. Oral versus intravenous high-dose methylprednisolone for treatment of relapses inpatients with multiple sclerosis (COPOUSEP): A randomised, controlled, double-blind, non-inferiority trial. Lancet 2015, 386, 974–981.
- Bonnan, M.; Cabre, P. Plasma Exchange in Severe Attacks of Neuromyelitis Optica. Mult. Scler. Int. 2012, 2012, 787630.
- Bennett, J.L. Optic Neuritis. Continuum 2019, 25, 1236–1264.
- Jakimovski, D.; Kolb, C.; Ramanathan, M.; Zivadinov, R.; Weinstock-Guttman, B. Interferon β for Multiple Sclerosis. Cold Spring Harb. Perspect. Med. 2018, 8, a032003.
- Dhib-Jalbut, S.; Marks, S. Interferon-beta mechanisms of action in multiple sclerosis. Neurology 2010, 74 (Suppl. S1), S17–S24.
- Rudick, R.A.; Ransohoff, R.M.; Lee, J.C.; Peppler, R.; Yu, M.; Mathisen, P.M.; Tuohy, V.K. In vivo effects of interferon beta-1a on immunosuppressive cytokines in multiple sclerosis. Neurology 1998, 50, 1294–1300.
- Hartrich, L.; Weinstock-Guttman, B.; Hall, D.; Badgett, D.; Baier, M.; Patrick, K.; Feichter, J.; Hong, J.; Ramanathan, M. Dynamics of immune cell trafficking in interferon-β treated multiple sclerosis patients. J. Neuroimmunol. 2003, 139, 84–92.
- Rizzo, F.; Giacomini, E.; Mechelli, R.; Buscarinu, M.C.; Salvetti, M.; Severa, M.; Coccia, E.M. Interferon-β therapy specifically reduces pathogenic memory B cells in multiple sclerosis patients by inducing a FAS-mediated apoptosis. Immunol. Cell. Biol. 2016, 94, 886–894.
- Molnarfi, N.; Prod’homme, T.; Schulze-Topphoff, U.; Spencer, C.M.; Weber, M.S.; Patarroyo, J.C.; Lalive, P.H.; Zamvil, S.S. Glatiramer acetate treatment negatively regulates type I interferon signaling. Neurol. Neuroimmunol. Neuroinflamm. 2015, 2, e179.
- Weber, M.S.; Hohlfeld, R.; Zamvil, S.S. Mechanism of action of glatiramer acetate in treatment of multiple sclerosis. Neurotherapeutics 2007, 4, 647–653.
- Prod’homme, T.; Zamvil, S.S. The Evolving Mechanisms of Action of Glatiramer Acetate. Cold Spring Harb. Perspect. Med. 2019, 9, a029249.
- Polman, C.H.; Reingold, S.C.; Banwell, B.; Clanet, M.; Cohen, J.A.; Filippi, M.; Fujihara, K.; Havrdova, E.; Hutchinson, M.; Kappos, L.; et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann. Neurol. 2011, 69, 292–302.
- Kieseier, B.C.; Benamor, M. Pregnancy outcomes following maternal and paternal exposure to teriflunomide during treatment for relapsing-remitting multiple sclerosis. Neurol. Ther. 2014, 3, 133–138.
- Andersen, J.B.; Moberg, J.Y.; Spelman, T.; Magyari, M. Pregnancy Outcomes in Men and Women Treated with Teriflunomide. A Population-Based Nationwide Danish Register Study. Front. Immunol. 2018, 9, 2706.
- Fazekas, F.; Lublin, F.D.; Li, D.; Freedman, M.S.; Hartung, H.P.; Rieckmann, P.; Sørensen, P.S.; Maas-Enriquez, M.; Sommerauer, B.; Hanna, K.; et al. Intravenous immunoglobulin in relapsing-remitting multiple sclerosis: A dose-finding trial. Neurology 2008, 71, 265–271.
- Sørensen, P.S.; Sellebjerg, F.; Lycke, J.; Färkkilä, M.; Créange, A.; Lund, C.G.; Schluep, M.; Frederiksen, J.L.; Stenager, E.; Pfleger, C.; et al. Minocycline added to subcutaneous interferon β-1a in multiple sclerosis: Randomized Recycline study. Eur. J. Neurol. 2016, 23, 861–870.
- Metz, L.M.; Li, D.K.B.; Traboulsee, A.L.; Duquette, P.; Eliasziw, M.; Cerchiaro, G.; Greenfield, J.; Riddehough, A.; Yeung, M.; Kremenchutzky, M.; et al. Trial of Minocycline in a Clinically Isolated Syndrome of Multiple Sclerosis. N. Engl. J. Med. 2017, 376, 2122–2133.
- Jacobs, L.D.; Beck, R.W.; Simon, J.H.; Kinkel, R.P.; Brownscheidle, C.M.; Murray, T.J.; Simonian, N.A.; Slasor, P.J.; Sandrock, A.W.; The CHAMPS Study Group. Intramuscular interferon beta-1a therapy initiated during a first demyelinating event in multiple sclerosis. N. Engl. J. Med. 2000, 343, 898–904.
- Goodin, D.S.; Bates, D. Treatment of early multiple sclerosis: The value of treatment initiation after a first clinical episode. Mult. Scler. 2009, 15, 1175–1182.
- Comi, G.; Filippi, M.; Barkhof, F.; Durelli, L.; Edan, G.; Fernández, O.; Hartung, H.; Seeldrayers, P.; Sørensen, P.S.; Rovaris, M.; et al. Effect of early interferon treatment on conversion to definite multiple sclerosis: A randomised study. Lancet 2001, 357, 1576–1582.
- Efendi, H. Clinically Isolated Syndromes: Clinical Characteristics, Differential Diagnosis, and Management. Noro Psikiyatr. Ars. 2015, 52 (Suppl. S1), S1–S11.
- Marcus, J.F.; Waubant, E.L. Updates on Clinically Isolated Syndrome and Diagnostic Criteria for Multiple Sclerosis. Neurohospitalist 2013, 3, 65–80.
- Gold, R.; Wolinsky, J.S. Pathophysiology of multiple sclerosis and the place of teriflunomide. Acta Neurol. Scand. 2011, 124, 75–84.
- Comi, G.; De Stefano, N.; Freedman, M.S.; Barkhof, F.; Polman, C.H.; Uitdehaag, B.M.J.; Casset-Semanaz, F.; Hennessy, B.; Moraga, M.S.; Rocak, S.; et al. Comparison of two dosing frequencies of subcutaneous interferon beta-1a in patients with a first clinical demyelinating event suggestive of multiple sclerosis (REFLEX): A phase 3 randomised controlled trial. Lancet Neurol. 2012, 11, 33.
- Comi, G.; De Stefano, N.; Freedman, M.S.; Barkhof, F.; Uitdehaag, B.M.; de Vos, M.; Marhardt, K.; Chen, L.; Issard, D.; Kappos, L. Subcutaneous interferon β-1a in the treatment of clinically isolated syndromes: 3-year and 5-year results of the phase III dosing frequency-blind multicentre REFLEXION study. J. Neurol. Neurosurg. Psychiatry 2017, 88, 285–294.
- Miller, A.E.; Wolinsky, J.S.; Kappos, L.; Comi, G.; Freedman, M.S.; Olsson, T.P.; Bauer, D.; Benamor, M.; Truffinet, P.; O’Connor, P.W.; et al. Oral teriflunomide for patients with a first clinical episode suggestive of multiple sclerosis (TOPIC): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Neurol. 2014, 13, 977–986.