 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Alfred Schinkel | -- | 3680 | 2022-09-16 14:42:54 | | | |
| 2 | Peter Tang | -35 word(s) | 3645 | 2022-09-19 03:17:51 | | |
Video Upload Options
Ritonavir is the most potent cytochrome P450 (CYP) 3A4 inhibitor in clinical use and is often applied as a booster for drugs with low oral bioavailability due to CYP3A4-mediated biotransformation, as in the treatment of HIV (e.g., lopinavir/ritonavir) and more recently COVID-19 (Paxlovid or nirmatrelvir/ritonavir). Ritonavir is clearly a potent mechanism-based inactivator, which irreversibly blocks CYP3A4.
1. Introduction
1.1. Properties of Cytochrome P450 Enzymes including CYP3A4 and CYP3A5
1.2. Ritonavir as a Clinically Important CYP3A Inhibitor

1.3. The Ritonavir Analogue Cobicistat Has Very Similar CYP3A Inhibition Properties
2. Binding of Ritonavir to CYP3A4 and CYP3A5
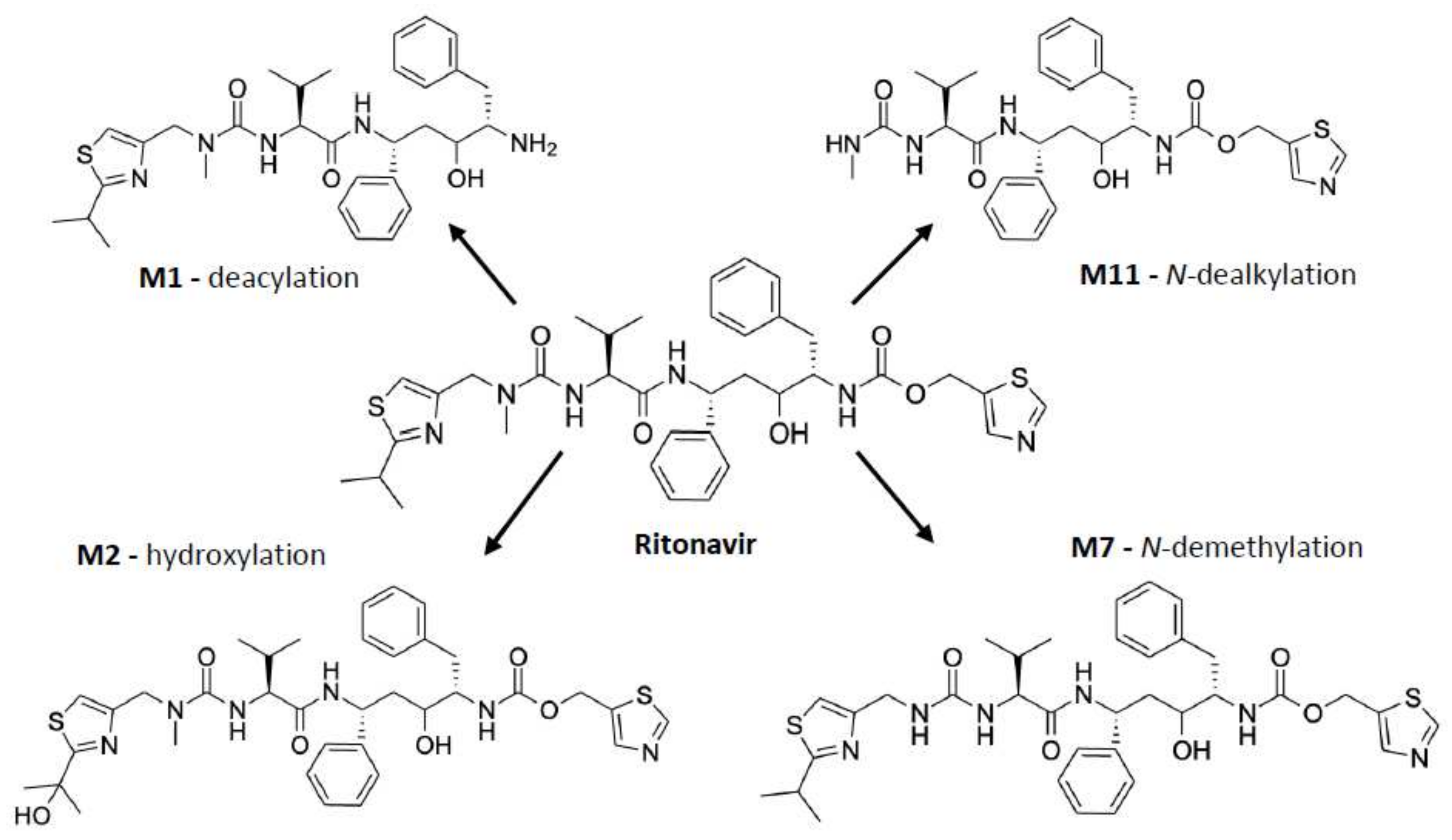
3. Metabolism of Ritonavir by CYP3A4 and -3A5
4. Principal Mechanisms of Irreversible Inhibition of CYP Enzymes by Substrates
|
Reference |
Suggested Primary Mechanism of Inactivation |
Used Enzyme Preparations |
Use of Added Cytb5 or CPR in the Experiments * |
Incubation Time |
Assay(s) |
Ritonavir Concentration |
|---|---|---|---|---|---|---|
|
Koudriakova et al. (1998) [24] |
Reactive intermediate formation |
Enterocyte microsomes and HLMs expressing CYP3A4, -3A5, and -2D6 |
- No Cytb5 - No CPR |
1 h |
Time-course assay using HPLC to examine the rate of ritonavir metabolism |
2 or 5 µM |
|
20 min |
Inactivation of CYP enzymes assay using HPLC |
0.075 µM |
||||
|
Ernest et al. (2005) [52] |
MIC formation |
HLMs expressing CYP3A4 and -3A5 |
- Recombinant CYP3A4: with Cytb5 - Recombinant CYP3A5: no Cytb5 - All with CPR |
Maximally 60 min |
CYP3A4/5 inactivation and high-affinity binding assay with testosterone substrate to quantify time- and concentration-dependent loss of CYP3A activity |
0.05, 0.10, 0.20, 0.50, and 1 µM |
|
Sevrioukova et al. (2010) [60] |
Strong ligation of ritonavir to heme iron |
Isolated CYP3A4Δ3-24 |
- No Cytb5 - No CPR |
- |
Kinetic assay of CYP3A4-ritonavir binding using stopped-flow spectrophotometry to measure the kinetics of ritonavir binding to ferric and ferrous P450 by monitoring absorbance changes at 426 and 442 nm |
0.5–30 μM |
|
Crystallization and structure determination with bound ritonavir |
Ritonavir-bound CYP3A4 protein (50–60 mg/mL) |
|||||
|
Lin et al. (2013) [62] |
Heme destruction and linkage of heme to apoprotein |
Purified CYP3A4 and CYP2B6, and HLMs |
- No Cytb5 - With CPR |
30 min |
Enzyme and inactivation assay of CYP3A4 and CYP2B6 to determine catalytic activity using a fluorescence plate reader |
0.5–20 µM |
|
10 min |
HPLC analysis of heme iron to study the loss of native heme and formation of heme adducts |
10 µM for CYP2B6; 2 µM for CYP3A4 |
||||
|
10 min |
ESI–LC/MS analysis of the apoprotein to study the mass spectra |
10 µM |
||||
|
20 min |
LC-MS/MS analysis of ritonavir metabolites and the GSH conjugate formed |
40 µM |
||||
|
Rock et al. (2014) [18] |
Reactive intermediate formation with covalent adduct binding to apoprotein (Lys257) |
CYP3A4 supersomes or HLMs |
- With Cytb5 - With CPR |
30 min, after 3 min pre-incubation |
CYP3A4 activity and inactivation assay using midazolam with a UPLC system and LC-MS/MS for the inactivation assay |
0–10 µM of ritonavir or N-ritonavir |
|
MIC formation assay using spectrophotometric repetitive scanning from 430–495 nm over 30 min |
10 µM |
|||||
|
With and without NADPH |
10 min |
Mass spectral analysis of CYP3A4 peptides using a liquid chromatography - radioisotope counting system |
10 µM |
* This column only contains the information of the experiments that were essential for the conclusions of the authors regarding their proposed mechanism of inactivation of CYP3A by ritonavir. In fact, the reviewed studies showed more detailed information including the experimental conditions. HLMs, human liver microsomes. CYP, cytochrome P450. Cytb5, cytochrome b5. CPR, cytochrome P450 reductase. NADPH, nicotinamide adenine dinucleotide phosphate. MIC, metabolic intermediate complex. GSH, glutathione. Lys257, lysine 257. HPLC, high-performance liquid chromatography. ESI-LC/MS, electrospray ionization mass spectrometry. LC-MS/MS, liquid chromatography with tandem mass spectrometry. UPLC, ultra-high-performance liquid chromatography.
References
- Lolodi, O.; Wang, Y.-M.; Wright, W.C.; Chen, T. Differential Regulation of CYP3A4 and CYP3A5 and its Implication in Drug Discovery. Curr. Drug Metab. 2018, 18, 1095–1105.
- Yadav, J.; Korzekwa, K.; Nagar, S. Improved Predictions of Drug–Drug Interactions Mediated by Time-Dependent Inhibition of CYP3A. Mol. Pharm. 2018, 15, 1979–1995.
- Thelen, K.; Dressman, J.B. Cytochrome P450-mediated metabolism in the human gut wall. J. Pharm. Pharmacol. 2009, 61, 541–558.
- Kim, J.H.; Sherman, M.E.; Curriero, F.C.; Guengerich, F.; Strickland, P.T.; Sutter, T.R. Expression of cytochromes P450 1A1 and 1B1 in human lung from smokers, non-smokers, and ex-smokers. Toxicol. Appl. Pharmacol. 2004, 199, 210–219.
- Ghosh, C.; Marchi, N.; Desai, N.K.; Puvenna, V.; Hossain, M.; Gonzalez-Martinez, J.; Alexopoulos, A.V.; Janigro, D. Cellular localization and functional significance of CYP3A4 in the human epileptic brain. Epilepsia 2011, 52, 562–571.
- Knops, N.; Heuvel, L.P.V.D.; Masereeuw, R.; Bongaers, I.; de Loor, H.; Levtchenko, E.; Kuypers, D. The Functional Implications of Common Genetic Variation in CYP3A5 and ABCB1 in Human Proximal Tubule Cells. Mol. Pharm. 2015, 12, 758–768.
- Van Eijk, M.; Boosman, R.J.; Schinkel, A.H.; Huitema, A.D.R.; Beijnen, J.H. Cytochrome P450 3A4, 3A5, and 2C8 expression in breast, prostate, lung, endometrial, and ovarian tumors: Relevance for resistance to taxanes. Cancer Chemother. Pharmacol. 2019, 84, 487–499.
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141.
- Krusekopf, S.; Roots, I.; Kleeberg, U. Differential drug-induced mRNA expression of human CYP3A4 compared to CYP3A5, CYP3A7 and CYP3A43. Eur. J. Pharmacol. 2003, 466, 7–12.
- Zhou, S.-F. Drugs Behave as Substrates, Inhibitors and Inducers of Human Cytochrome P450 3A4. Curr. Drug Metab. 2008, 9, 310–322.
- Ince, I.; Knibbe, C.A.J.; Danhof, M.; de Wildt, S.N. Developmental Changes in the Expression and Function of Cytochrome P450 3A Isoforms: Evidence from In Vitro and In Vivo Investigations. Clin. Pharmacokinet. 2013, 52, 333–345.
- Hsu, M.-H.; Savas, U.; Johnson, E.F. The X-ray Crystal Structure of the Human Mono-Oxygenase Cytochrome P450 3A5-Ritonavir Complex Reveals Active Site Differences between P450s 3A4 and 3A5. Mol. Pharmacol. 2018, 93, 14–24.
- Hsu, M.-H.; Johnson, E.F. Active-site differences between substrate-free and ritonavir-bound cytochrome P450 (CYP) 3A5 reveal plasticity differences between CYP3A5 and CYP3A4. J. Biol. Chem. 2019, 294, 8015–8022.
- Kuehl, P.; Zhang, J.; Lin, Y.; Lamba, J.; Assem, M.; Schuetz, J.; Watkins, P.B.; Daly, A.; Wrighton, S.A.; Hall, S.D.; et al. Sequence diversity in CYP3A promoters and characterization of the genetic basis of polymorphic CYP3A5 expression. Nat. Genet. 2001, 27, 383–391.
- Saito, Y.; Yamamoto, N.; Katori, N.; Maekawa, K.; Fukushima-Uesaka, H.; Sugimoto, D.; Kurose, K.; Sai, K.; Kaniwa, N.; Sawada, J.-I.; et al. Genetic Polymorphisms and Haplotypes of POR, Encoding Cytochrome P450 Oxidoreductase, in a Japanese Population. Drug Metab. Pharmacokinet. 2011, 26, 107–116.
- Yoo, S.-E.; Yi, M.; Kim, W.-Y.; Cho, S.-A.; Lee, S.S.; Lee, S.-J.; Shin, J.-G. Influences of cytochrome b5 expression and its genetic variant on the activity of CYP2C9, CYP2C19 and CYP3A4. Drug Metab. Pharmacokinet. 2019, 34, 201–208.
- Gan, L.; von Moltke, L.L.; Trepanier, L.A.; Harmatz, J.S.; Greenblatt, D.J.; Court, M.H. Role of NADPH-cytochrome P450 reductase and cytochrome-b5/NADH-b5 reductase in variability of CYP3A activity in human liver microsomes. Drug Metab. Dispos. 2009, 37, 90–96.
- Rock, B.M.; Hengel, S.M.; Rock, D.A.; Wienkers, L.C.; Kunze, K.L. Characterization of Ritonavir-Mediated Inactivation of Cytochrome P450 3A4. Mol. Pharmacol. 2014, 86, 665–674.
- Masubuchi, Y.; Horie, T. Toxicological Significance of Mechanism-Based Inactivation of Cytochrome P450 Enzymes by Drugs. Crit. Rev. Toxicol. 2007, 37, 389–412.
- Hu, B.; Zhou, X.; Mohutsky, M.A.; Desai, P.V. Structure–Property Relationships and Machine Learning Models for Addressing CYP3A4-Mediated Victim Drug–Drug Interaction Risk in Drug Discovery. Mol. Pharm. 2020, 17, 3600–3608.
- Jayakanthan, M.; Chandrasekar, S.; Muthukumaran, J.; Mathur, P.P. Analysis of CYP3A4-HIV-1 protease drugs interactions by computational methods for Highly Active Antiretroviral Therapy in HIV/AIDS. J. Mol. Graph. Model. 2010, 28, 455–463.
- Von Hentig, N.; Haberl, A. Safety of pharmacoenhancers for HIV therapy. Expert Rev. Clin. Pharmacol. 2012, 5, 557–568.
- Kempf, D.J.; Marsh, K.C.; Denissen, J.F.; McDonald, E.; Vasavanonda, S.; Flentge, C.A.; Green, B.E.; Fino, L.; Park, C.H.; Kong, X.P. ABT-538 is a potent inhibitor of human immunodeficiency virus protease and has high oral bioavailability in humans. Proc. Natl. Acad. Sci. USA 1995, 92, 2484–2488.
- Koudriakova, T.; Iatsimirskaia, E.; Utkin, I.; Gangl, E.; Vouros, P.; Storozhuk, E.; Orza, D.; Marinina, J.; Gerber, N. Metabolism of the human immunodeficiency virus protease inhibitors indinavir and ritonavir by human intestinal microsomes and expressed cytochrome P4503A4/3A5: Mechanism-based inactivation of cytochrome P4503A by ritonavir. Drug Metab. Dispos. 1998, 26, 552–561.
- Kumar, G.N.; Rodrigues, A.D.; Buko, A.M.; Denissen, J.F. Cytochrome P450-mediated metabolism of the HIV-1 protease inhibitor ritonavir (ABT-538) in human liver microsomes. J. Pharmacol. Exp. Ther. 1996, 277, 423–431.
- Margolis, A.M.; Heverling, H.; Pham, P.A.; Stolbach, A. A review of the toxicity of HIV medications. J. Med. Toxicol. 2014, 10, 26–39.
- Kageyama, M.; Namiki, H.; Fukushima, H.; Terasaka, S.; Togawa, T.; Tanaka, A.; Ito, Y.; Shibata, N.; Takada, K. Effect of Chronic Administration of Ritonavir on Function of Cytochrome P450 3A and P-Glycoprotein in Rats. Biol. Pharm. Bull. 2005, 28, 130–137.
- Fukushima, K.; Kobuchi, S.; Mizuhara, K.; Aoyama, H.; Takada, K.; Sugioka, N. Time-Dependent Interaction of Ritonavir in Chronic Use: The Power Balance between Inhibition and Induction of P-Glycoprotein and Cytochrome P450 3A. J. Pharm. Sci. 2013, 102, 2044–2055.
- Kirby, B.J.; Collier, A.C.; Kharasch, E.D.; Dixit, V.; Desai, P.; Whittington, D.; Thummel, K.E.; Unadkat, J.D. Complex Drug Interactions of HIV Protease Inhibitors 2: In Vivo Induction and In Vitro to In Vivo Correlation of Induction of Cytochrome P450 1A2, 2B6, and 2C9 by Ritonavir or Nelfinavir. Drug Metab. Dispos. 2011, 39, 2329–2337.
- Yeh, R.F.; Gaver, V.E.; Patterson, K.B.; Rezk, N.L.; Baxter-Meheux, F.; Blake, M.J.; Eron, J.J.; Klein, C.E.; Rublein, J.C.; Kashuba, A.D. Lopinavir/Ritonavir Induces the Hepatic Activity of Cytochrome P450 Enzymes CYP2C9, CYP2C19, and CYP1A2 But Inhibits the Hepatic and Intestinal Activity of CYP3A as Measured by a Phenotyping Drug Cocktail in Healthy Volunteers. JAIDS J. Acquir. Immune Defic. Syndr. 2006, 42, 52–60.
- Foisy, M.M.; Yakiwchuk, E.M.; Hughes, C.A. Induction Effects of Ritonavir: Implications for Drug Interactions. Ann. Pharmacother. 2008, 42, 1048–1059.
- Gupta, A.; Mugundu, G.M.; Desai, P.B.; Thummel, K.E.; Unadkat, J.D. Intestinal Human Colon Adenocarcinoma Cell Line LS180 Is an Excellent Model to Study Pregnane X Receptor, but Not Constitutive Androstane Receptor, Mediated CYP3A4 and Multidrug Resistance Transporter 1 Induction: Studies with Anti-Human Immunodeficiency Virus Protease Inhibitors. Drug Metab. Dispos. 2008, 36, 1172–1180.
- Fahmi, O.A.; Maurer, T.S.; Kish, M.; Cardenas, E.; Boldt, S.; Nettleton, D. A Combined Model for Predicting CYP3A4 Clinical Net Drug-Drug Interaction Based on CYP3A4 Inhibition, Inactivation, and Induction Determined In Vitro. Drug Metab. Dispos. 2008, 36, 1698–1708.
- Eagling, V.A.; Back, D.J.; Barry, M.G. Differential inhibition of cytochrome P450 isoforms by the protease inhibitors, ritonavir, saquinavir and indinavir. Br. J. Clin. Pharmacol. 1997, 44, 190–194.
- Von Moltke, L.L.; Greenblatt, D.J.; Grassi, J.M.; Granda, B.W.; Duan, S.X.; Fogelman, S.M.; Daily, J.P.; Harmatz, J.S.; Shader, R.I. Protease inhibitors as inhibitors of human cytochromes P450: High risk associated with ritonavir. J. Clin. Pharmacol. 1998, 38, 106–111.
- Zeldin, R.K.; Petruschke, R.A. Pharmacological and therapeutic properties of ritonavir-boosted protease inhibitor therapy in HIV-infected patients. J. Antimicrob. Chemother. 2004, 53, 4–9.
- Pasternak, A.O.; Vroom, J.; Kootstra, N.A.; Wit, F.W.; de Bruin, M.; De Francesco, D.; Bakker, M.; Sabin, C.A.; Winston, A.; Prins, J.M.; et al. Non-nucleoside reverse transcriptase inhibitor-based combination antiretroviral therapy is associated with lower cell-associated HIV RNA and DNA levels compared to protease inhibitor-based therapy. eLife 2021, 10, e68174.
- Carrillo, A.; Stewart, K.D.; Sham, H.L.; Norbeck, D.W.; Kohlbrenner, W.E.; Leonard, J.M.; Kempf, D.J.; Molla, A. In vitro selection and characterization of human immunodeficiency virus type 1 variants with increased resistance to ABT-378, a novel protease inhibitor. J. Virol. 1998, 72, 7532–7541.
- Highleyman, L. ABT-378: A second generation protease inhibitor. BETA 1998, 8, 55.
- Sham, H.L.; Kempf, D.J.; Molla, A.; Marsh, K.C.; Kumar, G.N.; Chen, C.-M.; Kati, W.; Stewart, K.; Lal, R.; Hsu, A.; et al. ABT-378, a Highly Potent Inhibitor of the Human Immunodeficiency Virus Protease. Antimicrob. Agents Chemother. 1998, 42, 3218–3224.
- Hsu, A.; Granneman, G.R.; Cao, G.; Carothers, L.; El-Shourbagy, T.; Baroldi, P.; Erdman, K.; Brown, F.; Sun, E.; Leonard, J.M. Pharmacokinetic interactions between two human immunodeficiency virus protease inhibitors, ritonavir and saquinavir. Clin. Pharmacol. Ther. 1998, 63, 453–464.
- Ahmad, B.; Batool, M.; Ain, Q.U.; Kim, M.S.; Choi, S. Exploring the Binding Mechanism of PF-07321332 SARS-CoV-2 Protease Inhibitor through Molecular Dynamics and Binding Free Energy Simulations. Int. J. Mol. Sci. 2021, 22, 9124.
- Macchiagodena, M.; Pagliai, M.; Procacci, P. Characterization of the non-covalent interaction between the PF-07321332 inhibitor and the SARS-CoV-2 main protease. J. Mol. Graph. Model. 2021, 110, 108042.
- Hammond, J.; Leister-Tebbe, H.; Gardner, A.; Abreu, P.; Bao, W.; Wisemandle, W.; Baniecki, M.; Hendrick, V.M.; Damle, B.; Simón-Campos, A.; et al. Oral Nirmatrelvir for High-Risk, Nonhospitalized Adults with COVID-19. N. Engl. J. Med. 2022, 386, 1397–1408.
- Yu, H.; Janssen, J.M.; Sawicki, E.; Van Hasselt, J.G.C.; De Weger, V.A.; Nuijen, B.; Schellens, J.H.M.; Beijnen, J.H.; Huitema, A.D.R. A Population Pharmacokinetic Model of Oral Docetaxel Coadministered with Ritonavir to Support Early Clinical Development. J. Clin. Pharmacol. 2020, 60, 340–350.
- De Weger, V.A.; Stuurman, F.E.; Koolen, S.L.W.; Moes, J.J.; Hendrikx, J.J.M.A.; Sawicki, E.; Thijssen, B.; Keessen, M.; Rosing, H.; Mergui-Roelvink, M.; et al. A Phase I Dose Escalation Study of Once-Weekly Oral Administration of Docetaxel as ModraDoc001 Capsule or ModraDoc006 Tablet in Combination with Ritonavir. Clin. Cancer Res. 2019, 25, 5466–5474.
- Greenblatt, D.J.; Harmatz, J.S. Ritonavir is the best alternative to ketoconazole as an index inhibitor of cytochrome P450-3A in drug-drug interaction studies. Br. J. Clin. Pharmacol. 2015, 80, 342–350.
- Darwich, A.S.; Aslam, U.; Ashcroft, D.M.; Rostami-Hodjegan, A. Meta-Analysis of the Turnover of Intestinal Epithelia in Preclinical Animal Species and Humans. Drug Metab. Dispos. 2014, 42, 2016–2022.
- Culmmerdek, K.; Vonmoltke, L.; Gan, L.; Horan, K.A.; Reynolds, R.; Harmatz, J.S.; Court, M.H.; Greenblatt, D.J.; Moltke, L.L. Effect of extended exposure to grapefruit juice on cytochrome P450 3A activity in humans: Comparison with ritonavir. Clin. Pharmacol. Ther. 2006, 79, 243–254.
- Katzenmaier, S.; Markert, C.; Riedel, K.-D.; Burhenne, J.; Haefeli, W.E.; Mikus, G. Determining the Time Course of CYP3A Inhibition by Potent Reversible and Irreversible CYP3A Inhibitors Using a Limited Sampling Strategy. Clin. Pharmacol. Ther. 2011, 90, 666–673.
- Knox, T.A.; Oleson, L.; von Moltke, L.L.; Kaufman, R.C.; Wanke, C.A.; Greenblatt, D.J. Ritonavir Greatly Impairs CYP3A Activity in HIV Infection with Chronic Viral Hepatitis. J. Acquir. Immune Defic. Syndr. 2008, 49, 358–368.
- Ernest, C.S., 2nd; Hall, S.D.; Jones, D.R. Mechanism-based inactivation of CYP3A by HIV protease inhibitors. J. Pharmacol. Exp. Ther. 2005, 312, 583–591.
- Von Moltke, L.L.; Durol, A.L.B.; Duan, S.X.; Greenblatt, D.J. Potent mechanism-based inhibition of human CYP3A in vitro by amprenavir and ritonavir: Comparison with ketoconazole. Eur. J. Clin. Pharmacol. 2000, 56, 259–261.
- Hossain, M.A.; Tran, T.; Chen, T.; Mikus, G.; Greenblatt, D.J. Inhibition of human cytochromes P450 in vitro by ritonavir and cobicistat. J. Pharm. Pharmacol. 2017, 69, 1786–1793.
- Sevrioukova, I.F.; Poulos, T.L. Ritonavir analogues as a probe for deciphering the cytochrome P450 3A4 inhibitory mechanism. Curr. Top. Med. Chem. 2014, 14, 1348–1355.
- Samuels, E.R.; Sevrioukova, I. Inhibition of Human CYP3A4 by Rationally Designed Ritonavir-like Compounds: Impact and Interplay of the Side Group Functionalities. Mol. Pharm. 2018, 15, 279–288.
- Mathias, A.A.; German, P.; Murray, B.P.; Wei, L.; Jain, A.; West, S.; Warren, D.; Hui, J.; Kearney, B.P. Pharmacokinetics and Pharmacodynamics of GS-9350: A Novel Pharmacokinetic Enhancer without Anti-HIV Activity. Clin. Pharmacol. Ther. 2010, 87, 322–329.
- Greenblatt, D.J. Antiretroviral boosting by cobicistat, a structural analog of ritonavir. Clin. Pharmacol. Drug Dev. 2014, 3, 335–337.
- Marzolini, C.; Gibbons, S.; Khoo, S.; Back, D. Cobicistat versus ritonavir boosting and differences in the drug–drug interaction profiles with co-medications. J. Antimicrob. Chemother. 2016, 71, 1755–1758.
- Sevrioukova, I.F.; Poulos, T.L. Structure and mechanism of the complex between cytochrome P4503A4 and ritonavir. Proc. Natl. Acad. Sci. USA 2010, 107, 18422–18427.
- Rendic, S. Summary of information on human CYP enzymes: Human P450 metabolism data. Drug Metab. Rev. 2002, 34, 83–448.
- Lin, H.-L.; D’Agostino, J.; Kenaan, C.; Calinski, D.; Hollenberg, P.F. The Effect of Ritonavir on Human CYP2B6 Catalytic Activity: Heme Modification Contributes to the Mechanism-Based Inactivation of CYP2B6 and CYP3A4 by Ritonavir. Drug Metab. Dispos. 2013, 41, 1813–1824.
- Li, F.; Lu, J.; Ma, X. Metabolomic Screening and Identification of the Bioactivation Pathways of Ritonavir. Chem. Res. Toxicol. 2011, 24, 2109–2114.
- Hsu, A.; Granneman, G.R.; Bertz, R.J. Erratum to Ritonavir: Clinical pharmacokinetics and interactions with other anti-HIV agents. Clin. Pharmacokinet. 1998, 35, 473.
- Kaspera, R.; Kirby, B.J.; Sahele, T.; Collier, A.C.; Kharasch, E.D.; Unadkat, J.D.; Totah, R.A. Investigating the contribution of CYP2J2 to ritonavir metabolism in vitro and in vivo. Biochem. Pharmacol. 2014, 91, 109–118.
- Lin, H.L.; Hollenberg, P.F. The inactivation of cytochrome P450 3A5 by 17alpha-ethynylestradiol is cytochrome b5-dependent: Metabolic activation of the ethynyl moiety leads to the formation of glutathione conjugates, a heme adduct, and covalent binding to the apoprotein. J. Pharmacol. Exp. Ther. 2007, 321, 276–287.
- Hollenberg, P.F.; Kent, U.M.; Bumpus, N.N. Mechanism-Based Inactivation of Human Cytochromes P450s: Experimental Characterization, Reactive Intermediates, and Clinical Implications. Chem. Res. Toxicol. 2008, 21, 189–205.
- Ho, H.K.; Chan, J.C.Y.; Hardy, K.D.; Chan, E.C.Y. Mechanism-based inactivation of CYP450 enzymes: A case study of lapatinib. Drug Metab. Rev. 2015, 47, 21–28.

