1. Introduction
1.1. Properties of Cytochrome P450 Enzymes including CYP3A4 and CYP3A5
Cytochrome P450 (CYP) enzymes form an essential superfamily of heme-containing monooxygenases that play an important role in the metabolism of countless endogenous and xenobiotic substances, including hormones, bile acids, and sterols, as well as many drugs
[1][2][1,2]. CYPs catalyze hydroxylation and oxidation reactions, generally leading to the higher hydrophilicity of their substrates. In the case of drugs, this can sometimes result in prodrug activation but more often results in the formation of molecular structures that are more suitable for further conjugation in the body such as glucuronidation, sulfation, or glutathionylation
[1]. These comparatively hydrophilic conjugates can generally be cleared more easily from the body than the parent drugs through excretion by the liver, small intestine, and kidneys. Although the liver is the major site of CYP-dependent drug metabolism, there are also variable levels of CYP expression in extrahepatic tissues, especially the small intestine, but also kidney, lung, and brain
[3][4][5][6][3,4,5,6]. Furthermore, CYP expression can occur in several tumor tissues
[7].
There are 57 CYP enzymes identified in humans, which are classified into 18 families, many of which have quite specific physiological functions
[8]. Of most relevance for drug detoxification are the human CYP1, CYP2, and CYP3 families, which metabolize approximately 80% of all clinically used drugs. The most important isoform of this subfamily is CYP3A4, which is abundant in the adult liver and small intestine and is responsible for the bulk of the drug-metabolic activities of the CYP3A subfamily
[1][9][1,10]. CYP3A4 activity is, therefore, an important determinant of the oral availability and overall systemic exposure of many of its substrate drugs. Since CYP3A4 activity can vary dramatically between but also within individuals due to its extensive inhibition or induction by a range of different drugs and food compounds, as well as by some genetic polymorphisms, this enzyme activity is a major cause of variable drug exposure in patients. This can lead to unpredictable underexposure (and thus under-treatment) but also lethal overexposure to a range of drugs
[1][10][1,11].
Another important member of this subfamily, CYP3A5, has about an 83% amino acid identity with CYP3A4 and demonstrates extensive overlapping but not identical substrate specificities
[11][12]. Judging from the 3D structures, most differences between the proteins are located in the helical F through G regions that form the flexible roof of the active site cavity and in the N-terminal region of the catalytic domain that shapes one side of the active pocket of CYP3A4
[12][13]. Consequently, the two enzymes have different active site structures: the active site of CYP3A5 is somewhat taller and narrower than that of CYP3A4
[12][13]. Some drugs are preferentially metabolized by either CYP3A4 or CYP3A5, and the exact nature or preponderance of the metabolites formed from shared substrates can differ between the two enzymes. CYP3A5 is relatively abundant in the kidneys and lungs and is polymorphically expressed in the adult liver and intestine
[1][9][1,10]. CYP3A5 expression and activity levels are highly dependent on the genotype and ethnicity of an individual; although lowly expressed in most Caucasians (primarily carrying the poorly splicing CYP3A5*3 allele), it is highly expressed in most sub-Saharan African ethnic groups (primarily carrying the CYP3A5*1 allele). Many Asian ethnic groups have intermediate frequencies of the CYP3A5*1 allele and thus functional CYP3A5 activity
[1][12][13][14][1,13,14,15]. For drugs that are preferentially metabolized by CYP3A5 over CYP3A4, the CYP3A5*1 genotype can, therefore, have an important impact on effective exposure to these drugs.
The monooxygenase reactions mediated by CYP3A4 and CYP3A5 are dependent on the interactions of the substrates with the heme iron atom. Generally speaking, these CYPs modify their substrates by using electrons received from NADPH through cytochrome P450 oxidoreductase (CPR) and/or cytochrome b5 (Cytb5) to reduce O
2 to H
2O and simultaneously generate an (often) hydroxylated or otherwise modified substrate
[15][16][17][16,17,18]. The initial binding of substrates and (most) inhibitors to CYPs displaces a water molecule coordinated to the heme iron and alters the absorption spectrum of the heme group. Depending on the nature of the substrate binding, it can induce a type I or type II bound/unbound difference absorption spectrum. Substrates and inhibitors binding directly to the heme iron generally induce type II difference spectra, usually scanned in a wavelength range between 410 and 550 nm. When reducing equivalents (electrons) are available, hydroxylation or modification of the bound substrate by CYP3A can proceed, consuming oxygen (O
2) and releasing water (H
2O) in the process
[17][18]. Spectrophotometric analysis of the heme group of CYP3As, especially around 450 nm, plays an important role in understanding their interaction with the substrates.
A striking feature of both CYP3A4 and CYP3A5 is their extremely broad substrate specificity, accommodating drug-like molecules with widely divergent structures and physicochemical properties, although many tend to be fairly hydrophobic. X-ray crystal structure analysis has revealed that this can be achieved because both enzymes have a large and flexible drug binding site. These binding sites can accommodate highly divergent substrate molecules, primarily using hydrophobic interactions. This, combined with the possibility of the induced fit of both the substrate molecule and the flexible peptide helices forming part of the drug binding site, can explain the extremely broad spectrum of drugs that are bound and metabolized by these proteins
[12][13][13,14].
One important consequence of the broad substrate specificity of CYP3A4 and -3A5, with the drug binding sites accommodating many different molecules, is that these enzymes are also susceptible to reversible and irreversible inhibition by a large range of coadministered drugs or food compounds. Unsurprisingly, often irreversible inhibition (or inactivation) has the largest impact. For the broader group of CYP enzymes, four main mechanisms of inactivation have been described: the (quasi-irreversible) coordination of the substrate to the prosthetic heme iron by the formation of a metabolic intermediate complex (MIC); the covalent binding of a formed reactive intermediate to the apoprotein of the enzyme; the direct alkylation of a reactive intermediate to the heme prosthetic group; or the destruction of the prosthetic heme group resulting in heme-derived fragments that could covalently alter the apoprotein of the enzyme
[10][18][19][11,19,20]. For some CYPs and inactivators, multiple mechanisms appear to apply side by side.
1.2. Ritonavir as a Clinically Important CYP3A Inhibitor
From a clinical perspective, CYP3A inhibition can be highly problematic, for example, it can unexpectedly enhance the exposure to a substrate drug, causing toxicity. However, inhibition can also be beneficial as quite a few drugs are so rapidly degraded by CYP3As that they do not reach or maintain therapeutic plasma levels. In such cases, CYP3A can be deliberately inhibited by the coadministration of a CYP3A inhibitor drug, for example, in HIV protease inhibitor booster regimens. Here, low-dose ritonavir (originally named ABT-538), which was originally developed as an HIV protease inhibitor, is often coadministered for its highly efficient CYP3A-inhibiting capacity
[10][20][21][22][23][11,21,22,23,24]. During the early development of ritonavir as an HIV protease inhibitor, several issues were encountered. Ritonavir had a poor systemic availability, mainly due to the high CYP3A-mediated first-pass effects, and additional efflux by P-glycoprotein (P-gp/ABCB1) to the bile and intestinal lumen
[23][24][25][24,25,26]. This poor oral availability required relatively high oral ritonavir doses of more than 1000 mg per day, resulting in a higher susceptibility to drug side effects, especially gastrointestinal adverse events
[26][27]. Moreover, this high dose of ritonavir led to the concurrent induction of several CYP enzymes, including CYP3A4, -1A2, -2B6, -2C9, and -2C19, and also the induction of drug transporters, such as P-gp and the breast cancer resistance protein (BCRP/ABCG2), and uridine diphosphate-glucuronosyltransferase (UGT)
[27][28][29][30][31][28,29,30,31,32]. This induction capacity of ritonavir is linked to the PXR and/or CAR signaling pathways
[29][32][30,33]. However, even during chronic ritonavir administration, its potent CYP3A inhibitory effect appears to predominate
[33][34].
The inhibitory capacity of ritonavir for CYP3A was first described by Eagling et al. (1997) and Von Moltke et al. (1998), reporting low in vitro K
i and IC
50 values of 0.019 µM and 0.034 µM, respectively, based on the inhibition of testosterone 6β-hydroxylation
[34][35][35,36]. As a result, it was realized that lower ritonavir doses could be used for its clinical application as a CYP3A inhibitor, namely 100–200 mg once or twice daily. These much lower doses reduced ritonavir’s side effects and caused much less enzyme induction, leading to markedly improved tolerability
[36][37]. Another complication of using ritonavir as a single protease inhibitor (as for other HIV protease inhibitors) was the rapid development of viral resistance and subsequent reduced therapeutic efficacy. As a consequence, modern combination HIV regimens were developed, often including at least one protease inhibitor as well as an NNRTI
[37][38].
A second-generation protease inhibitor, lopinavir (ABT-378), was developed to overcome some of the problems observed with ritonavir. Interestingly, lopinavir appeared to be rapidly metabolized by CYP3A and it was found that its oral availability could be dramatically boosted by the addition of ritonavir as a CYP3A inhibitor
[38][39][39,40]. The combination of both protease inhibitors, first described by Sham et al. (1998), led to the development of the first fixed-dose combination of ritonavir and lopinavir (Kaletra)
[40][41]. The use of ritonavir as a pharmacokinetic enhancer was further described in saquinavir drug–drug interaction studies
[41][42]. More recently, this same principle was applied for the approved oral COVID-19 drug Paxlovid, containing the SARS-CoV2-targeting protease inhibitor nirmatrelvir (PF-07321332), which is rapidly degraded by CYP3A. This drug is co-formulated with a low dose of ritonavir in order to slow down its CYP3A-mediated degradation and thus extend its half-life and therapeutic efficacy
[42][43][44][43,44,45].
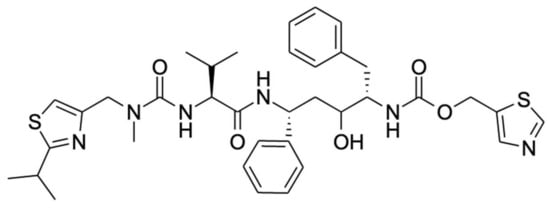
Ritonavir (
Figure 1) is one of the most efficacious inhibitors of CYP3A4/5 in routine clinical use and is often used to deliberately boost the oral availability of drugs that are otherwise extensively metabolized by CYP3A4/5
[10][11]. However, it is also an important clinical investigative and research tool to establish which (especially newly developed) drugs are likely to be extensively affected by CYP3A4/5-mediated metabolism
[45][46][46,47]. An important aspect of the clinical inhibition of CYP3A by ritonavir is its essentially irreversible nature in contrast with the efficient but fully reversible inhibition by compounds such as ketoconazole. Despite the reversible inhibitive nature of ketoconazole, the clinical CYP3A inactivation capacity of ritonavir appears only modestly higher in contrast with the inhibitory potency of itraconazole, which is significantly lower than that of ritonavir
[47][48]. As a consequence of the irreversible action, once CYP3A is inhibited by ritonavir in vivo, it will remain nonfunctional, and only its replacement with newly synthesized CYP3A will lead to a recurrence of CYP3A activity. The duration of the inhibition should thus to a large extent be dependent on the CYP3A turnover rate in the tissues in question, which might be quite rapid, especially in the small intestine, where entire human enterocytes have a turnover period of only about 3.5 days
[48][49]. Interestingly, there is no consensus in the literature about the clinical recovery time of CYP3A activity after the discontinuation of ritonavir. For example, a study by Culm-Merdek et al. (2006) described nearly full recovery after a three-day washout period, whereas Katzenmaier et al. (2011) observed the inactivity of the enzyme even after three days
[49][50][50,51]. The former results would appear to be more in line with the known turnover rate of enterocytes, but the replacement of CYP3A in hepatocytes could be considerably slower.
Figure 1.
Chemical structure of ritonavir.
Another important aspect of the long-term clinical use of ritonavir is that it does not seem to lose its boosting efficacy over time. Patients receiving ritonavir have sustained boosting effects despite extended periods of treatment, with little clinical evidence of induction of other CYP or UGT pathways
[51][52].
It is worth noting that although the time-dependent inhibition of CYP3A by ritonavir, that is, the mechanism-based inactivation, was first described in the late 1990s, it is now known that ritonavir also exhibits a significant reversible inhibitory capacity
[24][52][53][25,53,54]. As discussed later, ritonavir thus exhibits a mixed CYP3A inactivation potency
[54][55].
1.3. The Ritonavir Analogue Cobicistat Has Very Similar CYP3A Inhibition Properties
Interestingly, a close structural ritonavir analogue, cobicistat, is also registered (in 2014) and similar to other analogues is under further investigation for use as a clinical booster
[55][56][57][58][56,57,58,59]. Similar to ritonavir, cobicistat is a highly potent CYP3A inhibitor with a reversible and time-dependent component. The CYP3A IC
50 values of ritonavir and cobicistat are in the same range of 0.01–0.04 µM, although it seems that ritonavir has a slightly higher inhibitory potency compared to cobicistat. Nonetheless, the clinical effects as boosters are nearly identical for both ritonavir and cobicistat
[54][55]. According to the manufacturer, cobicistat should be more selective than ritonavir. However, Hossain et al. showed that this drug can also inhibit other CYP enzymes, including CYP2B6, CYP2C19, and CYP2D6, to possibly an even greater extent than ritonavir
[54][55]. In contrast, ritonavir also exhibits induction capacities through the PXR/CAR signaling pathways, whereas cobicistat appears to lack this ability to induce other CYP enzymes or transporters
[59][60]. Therefore, cobicistat could present a better drug–drug interaction profile than ritonavir.
2. Binding of Ritonavir to CYP3A4 and CYP3A5
Ritonavir was originally developed based on its structure/activity relationship with respect to inhibiting HIV protease, its intended molecular target. Its strong interaction with CYP3A4 is purely coincidental and not based on the CYP3A4 crystal structure, which was still unknown at the time
[55][56]. As discussed here, there are several theories about the primary mechanism of CYP3A inhibition or inactivation by ritonavir, which are not necessarily mutually exclusive. An important reason underlying the complexity in understanding this inhibition may be the flexibility and large size of the CYP3A drug binding site, allowing the induced fit of both the protein and/or its drug substrates and inhibitors. Moreover, multiple possible orientations and configurations of the substrates coordinating in the drug binding site, and even multiple substrates binding at the same time, can occur due to these properties
[8][60][61][8,61,62].
As shown in a comparison of X-ray crystal structures by Hsu et al., ritonavir exhibits higher configurational entropy when bound to CYP3A5 compared to CYP3A4, which is associated with quite different shapes of the active site cavity
[12][13]. Still, the binding affinity of ritonavir for these two enzymes is similar even though ritonavir adopts partly different conformations when bound by CYP3A4 or CYP3A5. Importantly, in these X-ray structures, the thiazole group of ritonavir shows an equally tight association with the heme iron in both enzymes. This, in combination with the high hydrophobicity and relatively poor aqueous solubility of ritonavir, may explain the high binding affinity to CYP3A4 and -3A5 rather than a strict lock and key fit model
[12][13][13,14]. The narrower active site of CYP3A5 leads to the redirection of the isopropyl-thiazole end of ritonavir to occupy an enlarged portion of the “roof” of the active site cavity above (and far away from) the heme
[12][13][13,14]. The active site of CYP3A5 shows reduced plasticity compared to CYP3A4 due to the differences in the width and height of their active cavities. Ritonavir shows higher configurational entropy with CYP3A5 because the molecule has some degree of freedom inside the active site. In contrast, the active site cavity of CYP3A4 widens and increases in height in the structure of the CYP3A4-ritonavir complex
[12][13]. This results in a highly restrained ritonavir molecule in this configuration of the CYP3A4 complex. Irrespective of the exact mechanism of binding and inhibition, experimentally it is clear that ritonavir is a potent (quasi-)irreversible inhibitor of both CYP3A4 and CYP3A5. However, due to the greater preponderance of CYP3A4 in the populations of most developed societies, ritonavir inhibition of CYP3A has so far been mostly investigated using CYP3A4.
3. Metabolism of Ritonavir by CYP3A4 and -3A5
Studies of the mechanism of inhibition of CYP3A by ritonavir are further complicated by the fact that CYP3A4 and CYP3A5 can also metabolize ritonavir to a range of different metabolites that are readily released from the proteins. Ritonavir is quite a complex, elongated molecule, containing 18 rotatable bonds allowing substantial structural flexibility (
Figure 1). It may well be that different orientations or configurations of ritonavir during entering into or upon positioning inside the equally flexible active sites of these proteins can either result in productive metabolism or irreversible inhibition.
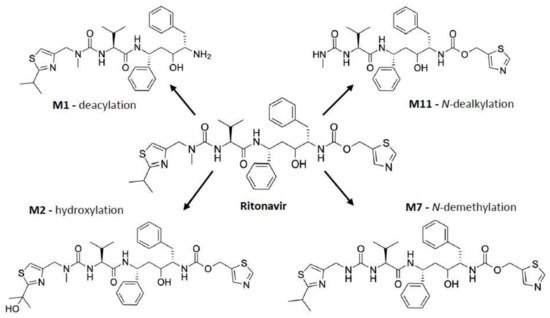
As shown in
Figure 2, ritonavir is primarily metabolized by human CYP3A4/5 through
N-demethylation, hydroxylation of the isopropyl side chain, and cleaving off of the terminal thiazole or isopropyl-thiazole groups, yielding four major metabolites amongst other metabolites formed during the biotransformation
[60][62][63][64][65][61,63,64,65,66]. Of note, the
N-demethylation reaction can also occur through human CYP2D6
[62][63]. The in vitro metabolism of ritonavir is characterized by an apparent
Km of 0.1–0.5 µM for recombinant CYP3A4. Moreover, the reaction is nonlinear, with a progressive slowing down of the metabolic conversion, presumably caused by the developing irreversible inhibition of CYP3A4
[60][61].
Figure 2. Ritonavir and its four major metabolites, which are M1 (deacylation), M2 (hydroxylation), and M11 (
N-dealkylation) formed by CYP3A4 and/or CYP3A5; and M7 (
N-demethylation) formed by CYP3A4/5, as well as CYP2D6
[62][65][63,66].
4. Principal Mechanisms of Irreversible Inhibition of CYP Enzymes by Substrates
Many different CYP enzymes are known to be irreversibly inactivated by substrate-like drugs or drug-like molecules, and this can happen through different primary inhibitory mechanisms. For CYP3A4/5, given the complexity of their substrates and the multiplicity of the chemical modifications they can make on the same substrate, multiple mechanisms at the same time might be relevant even for one drug. For instance, upon metabolic activation, the substrate 17-alpha-ethynylestradiol can irreversibly inactivate CYP3A5 by both heme modification and covalent binding to the apoprotein
[66][67]. The studies discussed here (
Table 1) variously propose four fundamentally different, irreversible inhibition mechanisms of CYP3A4/5 by ritonavir, namely the (I) formation of a true metabolic intermediate complex (MIC); (II) extremely tight binding of unchanged ritonavir to the heme iron; (III) heme destruction; and (IV) formation of a covalent bond with the CYP3A polypeptide by a reactive intermediate of ritonavir. Some of these currently proposed inhibitory mechanisms of CYP3A4/5 by ritonavir are referred to as the mechanism-based inactivation of the CYP3A4/5 enzyme. These are time-dependent and roughly correspond to the four different described types of mechanism-based inactivation for CYPs in general
[10][18][19][11,19,20]. Despite the differences between these proposed mechanisms of inactivation, the mechanism-based inactivators/inhibitors are generally considered to be compounds that are chemically converted by the target enzyme into a reactive intermediate or metabolic intermediate complex (MIC). This is then able to inactivate the enzyme by remaining attached to the active site or prior to its release from the active site cavity
[19][52][60][67][68][20,53,61,68,69].
Table 1.
Systematic overview of the divergent proposed mechanisms of mechanism-based inactivation of CYP3A by ritonavir and the used experimental approaches.
|
Reference
|
Suggested Primary Mechanism of Inactivation
|
Used Enzyme Preparations
|
Use of Added Cytb5 or CPR in the
Experiments *
|
Incubation Time
|
Assay(s)
|
Ritonavir
Concentration
|
|
Koudriakova et al. (1998) [24][25]
|
Reactive intermediate formation
|
Enterocyte microsomes and HLMs expressing CYP3A4, -3A5, and -2D6
|
- No Cytb5
- No CPR
|
1 h
|
Time-course assay using HPLC to examine the rate of ritonavir metabolism
|
2 or 5 µM
|
|
20 min
|
Inactivation of CYP enzymes assay using HPLC
|
0.075 µM
|
|
Ernest et al. (2005) [52][53]
|
MIC formation
|
HLMs expressing CYP3A4 and -3A5
|
- Recombinant CYP3A4: with Cytb5
- Recombinant CYP3A5: no Cytb5
- All with CPR
|
Maximally 60 min
|
CYP3A4/5 inactivation and high-affinity binding assay with testosterone substrate to quantify time- and
concentration-dependent loss of CYP3A activity
|
0.05, 0.10, 0.20, 0.50, and 1 µM
|
|
Sevrioukova et al. (2010) [60][61]
|
Strong ligation of ritonavir to heme iron
|
Isolated CYP3A4Δ3-24
|
- No Cytb5
- No CPR
|
-
|
Kinetic assay of CYP3A4-ritonavir binding using stopped-flow spectrophotometry to measure the kinetics of ritonavir binding to ferric
and ferrous P450 by monitoring absorbance changes at 426 and
442 nm
|
0.5–30 μM
|
|
Crystallization and structure determination with bound ritonavir
|
Ritonavir-bound CYP3A4
protein (50–60 mg/mL)
|
|
Lin et al. (2013) [62][63]
|
Heme destruction and linkage of heme to apoprotein
|
Purified CYP3A4 and CYP2B6, and HLMs
|
- No Cytb5
- With CPR
|
30 min
|
Enzyme and inactivation assay of CYP3A4 and CYP2B6 to determine catalytic activity using a fluorescence plate reader
|
0.5–20 µM
|
|
10 min
|
HPLC analysis of heme iron to study the loss of native heme and formation
of heme adducts
|
10 µM for CYP2B6; 2 µM for CYP3A4
|
|
10 min
|
ESI–LC/MS analysis of the apoprotein to study the mass spectra
|
10 µM
|
|
20 min
|
LC-MS/MS analysis of ritonavir metabolites and the GSH conjugate formed
|
40 µM
|
|
Rock et al. (2014) [18][19]
|
Reactive intermediate formation with covalent adduct binding to apoprotein (Lys257)
|
CYP3A4 supersomes or HLMs
|
- With Cytb5
- With CPR
|
30 min, after 3 min pre-incubation
|
CYP3A4 activity and inactivation assay using midazolam with a UPLC system and LC-MS/MS for the inactivation assay
|
0–10 µM of ritonavir or N-ritonavir
|
|
MIC formation assay using spectrophotometric
repetitive scanning from 430–495 nm over 30 min
|
10 µM
|
|
With and without NADPH
|
10 min
|
Mass spectral analysis of CYP3A4 peptides using a liquid chromatography -
radioisotope counting system
|
10 µM
|
* This column only contains the information of the experiments that were essential for the conclusions of the authors regarding their proposed mechanism of inactivation of CYP3A by ritonavir. In fact, the reviewed studies showed more detailed information including the experimental conditions. HLMs, human liver microsomes. CYP, cytochrome P450. Cytb5, cytochrome b5. CPR, cytochrome P450 reductase. NADPH, nicotinamide adenine dinucleotide phosphate. MIC, metabolic intermediate complex. GSH, glutathione. Lys257, lysine 257. HPLC, high-performance liquid chromatography. ESI-LC/MS, electrospray ionization mass spectrometry. LC-MS/MS, liquid chromatography with tandem mass spectrometry. UPLC, ultra-high-performance liquid chromatography.