Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Caterina Scuderi | -- | 2550 | 2022-09-16 14:28:51 | | | |
| 2 | Conner Chen | -63 word(s) | 2487 | 2022-09-20 05:26:03 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Valenza, M.; Facchinetti, R.; Steardo, L.; Scuderi, C. Palmitoylethanolamide and White Matter Lesions. Encyclopedia. Available online: https://encyclopedia.pub/entry/27252 (accessed on 10 August 2026).
Valenza M, Facchinetti R, Steardo L, Scuderi C. Palmitoylethanolamide and White Matter Lesions. Encyclopedia. Available at: https://encyclopedia.pub/entry/27252. Accessed August 10, 2026.
Valenza, Marta, Roberta Facchinetti, Luca Steardo, Caterina Scuderi. "Palmitoylethanolamide and White Matter Lesions" Encyclopedia, https://encyclopedia.pub/entry/27252 (accessed August 10, 2026).
Valenza, M., Facchinetti, R., Steardo, L., & Scuderi, C. (2022, September 16). Palmitoylethanolamide and White Matter Lesions. In Encyclopedia. https://encyclopedia.pub/entry/27252
Valenza, Marta, et al. "Palmitoylethanolamide and White Matter Lesions." Encyclopedia. Web. 16 September, 2022.
Copy Citation
Palmitoylethanolamide (PEA), the naturally occurring amide of ethanolamine and palmitic acid, is an endogenous lipid compound endowed with a plethora of pharmacological functions, including analgesic, neuroprotective, immune-modulating, and anti-inflammatory effects. The ability of formulations containing PEA in promoting oligodendrocyte differentiation, which represents the first step for the proper formation of myelin. This evidence opens new and promising research opportunities. White matter defects have been detected in a vast and heterogeneous group of diseases, including age-related neurodegenerative disorders.
palmitoylethanolamide
luteolin
oligodendrocyte
1. How Nature Provides Therapeutic Molecules: The History of Palmitoylethanolamide
The discovery of palmitoylethanolamide (PEA) arises from an interesting clinical observation in the early 1940s, when some clinicians reported that adding dried chicken egg yolk to the diets of underprivileged children reduced recurrences of rheumatic fever, despite the presence of streptococcal infections [1]. Later on, anti-allergic effects of the lipid fractions purified from egg yolk [2][3], peanut oil, and soybean lecithin [4] were demonstrated in guinea pigs. The agent responsible for these properties was definitively recognized as PEA [3], which was also identified in mammalian tissues fifteen years later [5].
The following years witnessed a series of interesting clinical studies on this molecule with good results. During the late 1970s, PEA clinical use was approved in former Czechoslovakia for acute respiratory diseases. For unknown reasons, not related to its toxicity, PEA was withdrawn from clinical use after several years on the market. In fact, the molecule remained largely unnoticed by the rest of the world for more than 20 years. Indeed, the market withdrawal and the failure to identify PEA molecular targets caused a period of research stasis. In the 1990s, Nobel laureate Prof. Rita Levi-Montalcini demonstrated that PEA is an endogenously produced regulator of inflammation [6]. This evidence, together with the discovery of the endocannabinoid system, caused a renewal of interest in PEA. In particular, during the last two decades, formulations containing PEA have received increasing attention as drugs and dietary supplements to be used in chronic pain, inflammation, and certain brain diseases.
2. The Pharmacology of Palmitoylethanolamide
PEA exhibits a rather complex biological and pharmacological profile. Despite some structural and metabolic similarities with the endocannabinoid signaling molecules, PEA is not a classical cannabinoid. It is occasionally called a “promiscuous” molecule, as it binds several receptors and also interacts with non-receptor targets (Figure 1). This alleged lack of selectivity is, however, increasingly regarded as an advantage in certain diseases, where “multiple-target” molecules may exert more beneficial effects than classical “single-target” drugs [7].
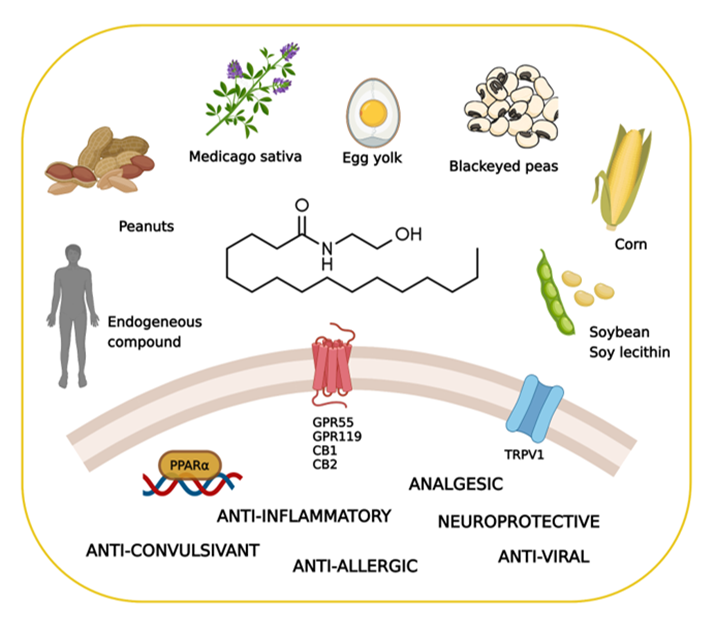
Figure 1. Key facts on palmitoylethanolamide (PEA), including its main sources, molecular targets, and effects. Created with BioRender.com (accessed on 22 July 2022).
2.1. Biosynthesis, Degradation, and Pharmacokinetics of PEA
PEA is an endogenous compound belonging to the family of N-acylethanolamines (NAEs) which include the endogenous cannabinoid receptor ligand anandamide (AEA, arachidonoylethanolamide) and the satiety agent oleoylethanolamide (OEA) [8][9].
Biosynthesis of PEA occurs on-demand within the lipid bilayer in two steps [10]. The first is the calcium- and cAMP-dependent transfer of palmitic acid from phosphatidylcholine to phosphatidylethanolamine to form N-acylphosphatidylethanolamine (NAPE); the second step is the cleavage of NAPE to release PEA, mediated by a NAPE-specific phospholipase D. The inactivation of PEA occurs via fatty acid amide amidohydrolase (FAAH) or PEA-preferring acid amidase (PAA) to form palmitic acid and ethanolamine.
All tissues, including the brain, have been found to contain PEA [11]. The physiological regulation of PEA levels in mammalian tissues is still under investigation. Different studies suggest that this compound accumulates after tissue injury, causing cellular stress; this supports the hypothesis that PEA is an endogenous mediator whose levels increase to exert a local reparative action [12].
Exogenous PEA administration presents some issues regarding its bioavailability. Given its lipid nature, PEA solubility in most aqueous solvents is indeed very low. It was shown that after intraperitoneal administration of radio-labeled-PEA to rats, the molecule was distributed mainly in some peripheral organs, whereas only low concentrations were detected in the brain and plasma [13]. Other groups demonstrated the ability of PEA to cross the blood–brain barrier (BBB) after oral administration, but only in very small amounts [14][15]. Of note, one clinical study reported that, in a dose-dependent manner, PEA administration leads to a two- to nine-fold increase in PEA plasma baseline concentrations [16].
To overcome the poor pharmacokinetics of PEA, different formulations containing PEA as micronized or ultra-micronized particles, or as ester derivatives (prepared by conjugating PEA with various amino acids), have been recently developed. These formulations improve PEA bioavailability in the central nervous system (CNS), without affecting its pharmacological efficacy [8].
2.2. The Search for the Palmitoylethanolamide Receptor
Between the 1950s and the 1980s, the mechanism(s) of action of PEA remained unidentified. This probably caused the lack of interest of the scientific community in PEA, despite its potential clinical significance. The renaissance of PEA originates from the work of Prof. Levi-Montalcini, who suggested that this endogenous compound exerts anti-inflammatory effects by serving as an autacoid local injury antagonist (ALIA) leading to a down-regulation of mast cell activation [6]. Moreover, the concurrent discovery of the endocannabinoid AEA and the cannabinoid receptors CB1 and CB2 shed new interest in PEA. Indeed, the similarity in chemical structure between AEA and PEA first suggested that these two endogenous mediators might share the same receptor. Several preclinical studies have definitely clarified that PEA does not bind the CB receptors and identified other mechanisms of action. The main molecular target of PEA seems to be the peroxisome proliferator-activated receptor-alpha (PPAR-α), through which PEA exerts its strong anti-inflammatory effects. Supporting this evidence, PEA effects vanished in mutant PPAR-α-null mice [10]. In particular, PPAR-α emerged as the key factor in mediating the ability of PEA to control neuroinflammation in different brain diseases [17][18][19][20][21][22][23][24].
Additionally, PEA can directly activate the G protein-coupled receptor 55 (GPR55) [25] and G protein-coupled receptor 119 (GPR119) [11]. Furthermore, it has been proposed that PEA indirectly potentiates the CB1 signal by inhibiting the degradation of AEA, a phenomenon known as the “entourage effect” [26][27]. Additionally, several studies have demonstrated the involvement of the transient receptor potential vanilloid 1 (TRPV1) in the actions of PEA [28][29][30][31]. The main PEA targets are schematized in Figure 1.
3. Myelin Sheath Organization and Functions
The term myelin was coined by Rudolf Virchow in 1864 and derives from the Greek word “myeloid” (marrow). Myelin provides the structural basis for fast impulse propagation along neuronal axons, a process required for the proper performance of motor, sensory, and cognitive functions in the CNS [32]. The myelin sheath is an extension of the plasma membrane of mature oligodendrocytes, cells belonging to the family of glial cells [33]. Oligodendrocytes originate from oligodendrocyte precursor cells (OPCs). During development, OPCs migrate from the ventricular/subventricular zones of the CNS to the whole brain, where they proliferate [33]. Differentiation into oligodendrocytes was the first acknowledged function of OPCs [34]. However, a rather large percentage of OPCs never differentiate and live throughout adulthood, suggesting that OPCs may play additional important roles [35]. For instance, in adulthood, when a myelin lesion occurs (e.g., in cases of brain damage), local OPCs begin to proliferate and differentiate into myelinating oligodendrocytes [34][36].
The maturation of OPCs requires several stages, each characterized by distinctive morphological and functional aspects and by the expression of specific proteins (Figure 2).
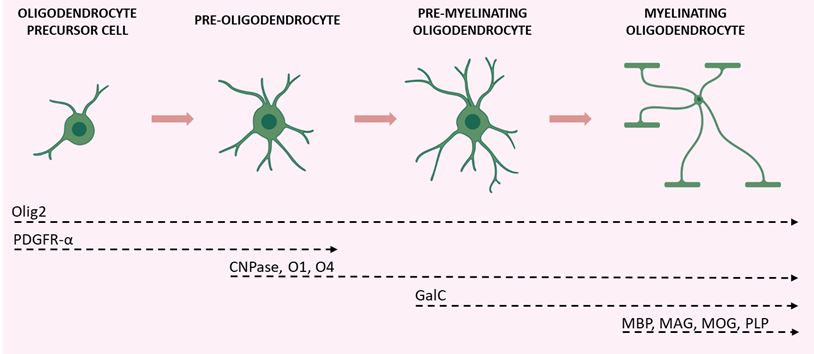
Figure 2. Schematic illustration of the various stages of OPC maturation and corresponding markers. Olig2 is expressed in all stages of the lineage; OPCs and pre-oligodendrocytes are characterized by PDGFR-α expression; CNPase, O1, and O4 are expressed during transition from pre-oligodendrocytes to differentiated oligodendrocytes (which also express GalC); axon-myelinating oligodendrocytes are characterized by myelin protein expression (MBP, MAG, MOG, and PLP). Created with BioRender.com (accessed on 19 August 2022).
Early OPCs are bipolar cells characterized mainly by the expression of the platelet-derived growth factor receptor-α (PDGFR-α). This receptor mediates the signal of the neuronal and astrocytic PDGF-α, which regulates the proliferation, migration, survival, and maturation of OPCs [37]. Once these cells keep contact with a target axon, they lose their bipolarity and start to develop filamentous myelin outgrowths, developing a complex shape. At this differentiation stage, pre-oligodendrocytes express 2′, 3′-cyclic-nucleotide 3′-phosphodiesterase (CNPase) and the cell surface markers O4 and O1. Finally, once fully mature, oligodendrocytes show a complex and ramified structure and form myelin sheaths. Myelinating oligodendrocytes can be identified by the expression of several markers including the myelin basic protein (MBP), the proteolipid protein (PLP), and the myelin-associated glycoprotein (MAG), as well as by the membrane marker galactocerebroside (GalC) and the surface marker myelin-oligodendrocyte glycoprotein (MOG) [38]. Olig2 is a transcription factor that drives oligodendrocyte-lineage cells to express myelin-related genes. It appears to be constantly expressed by oligodendrocytes regardless of the degree of differentiation, but progressively decreases at maturity [39].
The myelin structure consists of a multilayered battery of membranes organized in alternating electron-dense and electron-light layers [40]. The electron-dense layers represent the zone of adhesion between the tightly condensed cytoplasmic membranes, while the electron-light layers consist of extracellularly arranged myelin membranes [40]. The myelinated portions along axons are separated by the nodes of Ranvier, small myelin-free areas that promote rapid saltatory transmission of the nerve impulse by concentrating voltage-dependent sodium channels [41]. Myelin structure is stabilized by a multitude of adhesion mechanisms and proteins. The compaction of myelin is primarily performed by MBP, which condensates two adjacent cytoplasmic membrane surfaces into a major dense line of myelin [42][43]. The compaction begins in the very outer layers, close to the cell soma, and proceeds to the inner side in parallel with the growth of the membrane. In order to ensure the success of this process, MBP mRNA is trafficked from the nucleus to the myelin compartment where it is locally translated [44]. The integral membrane protein PLP allows the extracellular and cytoplasmic membranes to adhere to each other, enhancing the compaction of myelin [45]. The presence of the cytoplasm-rich compartments within myelin is ensured by CNPase which, by directly associating with cytoskeletal actin, antagonizes the adhesive forces exerted by polymerizing MBP [46]. Cytoplasmic channels allow the bidirectional movement of macromolecules under the myelin sheath maintaining vital such a compacted structure.
Through such channels, oligodendrocytes provide neurons with trophic and metabolic support. For example, oligodendrocytes use monocarboxylate transporters to deliver energy metabolites, like pyruvate and lactate, to neurons that use them to produce ATP [47].
4. The Role of the Astrocyte-Oligodendrocyte Cross-Talk in Myelination
The proliferation and differentiation of OPCs need the support of other glial cells, mainly microglia and astrocytes that release distinct patterns of secreted molecules to drive these processes. Microglia involvement in OPC differentiation, as well as in the context of myelin repair, is acknowledged, and the data in the literature highlight a dual role of these cells, providing evidence for both trophic and detrimental roles of microglia on oligodendrocytes and myelin (for a comprehensive review see [48]). Likewise, the role of astrocytes in the proliferation of OPCs and the formation and repair of myelin has long been recognized. It is believed that impairments of these star-shaped cells are implicated in the development of demyelinating diseases. This was first hypothesized by Müller in 1904, who was convinced that multiple sclerosis (MS), a demyelinating disease, was characterized by astrocytic dysfunction [49]. Research findings collected so far have confirmed this hypothesis, with various studies highlighting the key role of the cross-talk between astrocytes and oligodendrocytes in myelination, in both health and disease [50][51].
Astrocytes are the most abundant glial cell type of the CNS, found in both white and grey matter [52]. They were long considered secondary to neurons and were defined, from the Greek term glia, as “brain glue”. However, research studies over the past two decades have demonstrated that astrocytes exert a plethora of different functions to maintain CNS homeostasis at molecular, cellular, organ, and system levels of organization [53]. Astrocytes provide both physical and metabolic support to neurons [50] and modulate synaptic transmission and information processing by neural circuits [54]. Astrocytes are key components of the BBB; in this way, they regulate cerebral blood flow and the communication between the CNS and the periphery [54]. Furthermore, they are involved in several processes, such as ion and water transport, pH buffering, neuroplasticity, and synapse pruning. They also release approximately 200 molecules, including neurotrophic factors and energy substrates, thus, providing trophic and metabolic support to all cells in the CNS [55].
Astrocytes and oligodendrocytes originate from a common lineage of neural progenitor cells within the neuroectoderm [56] and, after development, communicate in physical and functional ways. Physically, astrocytes are coupled to oligodendrocytes through gap junctions at the cell body level and the paranodes, directly connecting the outer layer of the myelin sheath with an astrocytic process [57][58]. Such intercellular channels are heterologous, since oligodendrocytes express connexin (Cx) 32, Cx47, and Cx29, whereas astrocytes have Cx26, Cx30, and Cx43 [59]. A large number of reports underscore the importance of gap junctions in myelination. For instance, mutations or genetic ablation of specific connexins could lead to myelin defects, demyelinating diseases, encephalopathies, and peripheral neuropathies [60].
Besides direct physical interaction, astrocytes can communicate with oligodendrocytes by releasing a variety of soluble factors, including PDGF, brain-derived neurotrophic factor (BDNF), ciliary neurotrophic factor (CNTF), transforming growth factor (TGF)-β, and basic fibroblast growth factor (FGF2). All of these are considered promoters of OPC proliferation and survival [48], despite some contrasting evidence suggesting that PDGF and FGF2 could also act as inhibitors [61][62]. Moreover, the tissue inhibitor of metalloproteinase-1 (TIMP-1), an endogenous regulator of matrix metalloproteinases, promotes oligodendrogenesis [63]. In addition, astrocytes maintain and support myelin sheath formation by synthesizing and delivering cholesterol to oligodendrocytes [64][65].
As astrocytes are critical for maintaining brain homeostasis [50], they promptly react to any CNS insult or damage by undergoing morphofunctional changes that are context-, time-, and disease-specific [66][67][68][69]. Astrocyte reactivity could have either beneficial or detrimental consequences for myelination, and the difference seems to be related to the severity of astrogliosis [70]. Mild astrogliosis leads to the release of CNTF, FGF2, and the proinflammatory interleukin (IL)-6, all associated with OPC survival, proliferation, and maturation [71][72]. During severe astrogliosis, astrocytes secrete tumor necrosis factor (TNF)-α, which correlates with the extent of demyelination in MS and with myelin and oligodendrocytes damage in vitro [73][74]. Additionally, in an animal model of experimental autoimmune encephalomyelitis (EAE) the release of interferon (IFN)-γ was shown to suppress remyelination and delay recovery [75].
The relevance of astrocyte contribution to oligodendrocyte function is also evident in several astrocytopathies, such as Alexander disease, vanishing white matter, megalencephalic leukoencephalopathy with subcortical cysts, Aicardi–Goutières syndrome, and oculodentodigital dysplasia [76]. All these diseases are characterized by genetic mutations that cause defective astrocyte function, with major consequences for oligodendrocyte physiology as low oligodendrocyte survival, impaired myelination, and absence of myelin with or without concurrent development of astrogliosis [77][78].
Based on this evidence, astrocytes appear as the major cells orchestrating cell-to-cell communication relevant for myelination. Thus, alterations in this cross-talk could lead to myelin defects (Figure 3).
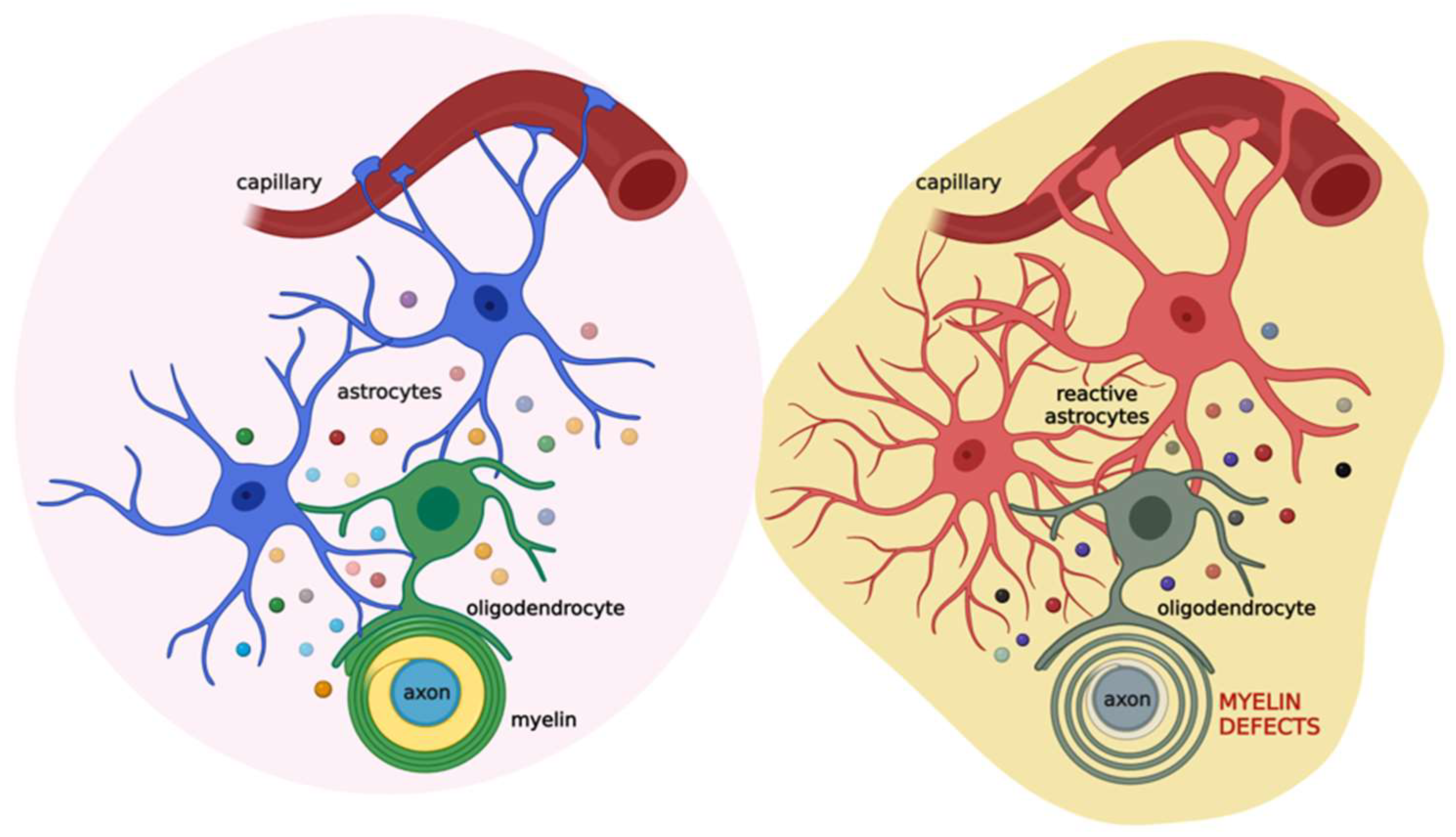
Figure 3. Schematic illustration of the coupling of oligodendrocytes, astrocytes, and neurons in physiological (left) and pathological (right) conditions. Astrocytes (blue) communicate with oligodendrocytes (green) through gap junctions and several released factors (colored circles), such as PDGF, BDNF, CNTF, TGF-β, FGF2, TIMP-1, ILs, TNF-α, and IFN-γ. This cross-talk allows the proper maturation of oligodendrocytes and their ability to form the myelin sheath. This, in turn, impacts neuronal activity. When brain damage occurs (see text for details), astrocytes (red) react promptly, affecting oligodendrocytes (grey) activity and ultimately the myelin sheath. Created with BioRender.com (accessed on 22 July 2022).
References
- Coburn, A.; Moore, L. American Journal of Diseases of Children. JAMA Pediatrics 1943, 65, 744–756.
- Coburn, A.F.; Graham, C.E.; Haninger, J. The effect of egg yolk in diets on anaphylactic arthritis (passive Arthus phenomenon) in the guinea pig. J. Exp. Med. 1954, 100, 425–435.
- Long, D.A.; Martin, A.J. Factor in arachis oil depressing sensitivity to tuberculin in B.C.G.-infected guineapigs. Lancet 1956, 270, 464–466.
- Long, D.; Miles, A. Opposite actions of thyroid and adrenal hormones in allergic hypersensitivity. Lancet 1950, 255, 492–495.
- Bachur, N.R.; Masek, K.; Melmon, K.L.; Udenfriend, S. Fatty Acid Amides of Ethanolamine in Mammalian Tissues. J. Biol. Chem. 1965, 240, 1019–1024.
- Aloe, L.; Leon, A.; Levi-Montalcini, R. A proposed autacoid mechanism controlling mastocyte behaviour. Agents Actions 1993, 39, C145–C147.
- Valenza, M.; Facchinetti, R.; Menegoni, G.; Steardo, L.; Scuderi, C. Alternative Targets to Fight Alzheimer’s Disease: Focus on Astrocytes. Biomolecules 2021, 11, 600.
- Beggiato, S.; Tomasini, M.C.; Ferraro, L. Palmitoylethanolamide (PEA) as a Potential Therapeutic Agent in Alzheimer’s Disease. Front. Pharm. 2019, 10, 821.
- Romano, A.; Di Bonaventura, M.V.M.; Gallelli, C.A.; Koczwara, J.B.; Smeets, D.; Giusepponi, M.E.; De Ceglia, M.; Friuli, M.; Di Bonaventura, E.M.; Scuderi, C.; et al. Oleoylethanolamide decreases frustration stress-induced binge-like eating in female rats: A novel potential treatment for binge eating disorder. Neuropsychopharmacology 2020, 45, 1931–1941.
- Lo Verme, J.; Fu, J.; Astarita, G.; La Rana, G.; Russo, R.; Calignano, A.; Piomelli, D. The nuclear receptor peroxisome proliferator-activated receptor-alpha mediates the anti-inflammatory actions of palmitoylethanolamide. Mol. Pharm. 2005, 67, 15–19.
- Rankin, L.; Fowler, C.J. The Basal Pharmacology of Palmitoylethanolamide. Int. J. Mol. Sci. 2020, 21, 7942.
- Darmani, N.A.; Izzo, A.A.; Degenhardt, B.; Valenti, M.; Scaglione, G.; Capasso, R.; Sorrentini, I.; Di Marzo, V. Involvement of the cannabimimetic compound, N-palmitoyl-ethanolamine, in inflammatory and neuropathic conditions: Review of the available pre-clinical data, and first human studies. Neuropharmacology 2005, 48, 1154–1163.
- Zhukov, O.D. -palmitoyl)ethanolamine in rat tissues]. Ukr. Biokhimichnyi Zhurnal 1999, 71, 124–125.
- Artamonov, M.; Zhukov, O.; Shuba, I.; Storozhuk, L.; Khmel, T.; Klimashevsky, V.; Mikosha, A.; Gula, N. Incorporation of labelled N-acylethanolamine (NAE) into rat brain regions in vivo and adaptive properties of saturated NAE under X-ray irradiation. Ukr. Biokhimichnyi Zhurnal 2005, 77, 51–62.
- Gabrielsson, L.; Mattsson, S.; Fowler, C.J. Palmitoylethanolamide for the treatment of pain: Pharmacokinetics, safety and efficacy. Br. J. Clin. Pharm. 2016, 82, 932–942.
- Balvers, M.G.; Verhoeckx, K.C.; Meijerink, J.; Wortelboer, H.M.; Witkamp, R.F. Measurement of palmitoylethanolamide and other N-acylethanolamines during physiological and pathological conditions. CNS Neurol. Disord. Drug Targets 2013, 12, 23–33.
- Straus, D.S.; Glass, C.K. Anti-inflammatory actions of PPAR ligands: New insights on cellular and molecular mechanisms. Trends Immunol. 2007, 28, 551–558.
- Scuderi, C.; Stecca, C.; Valenza, M.; Ratano, P.; Bronzuoli, M.R.; Bartoli, S.; Steardo, L.; Pompili, E.; Fumagalli, L.; Campolongo, P.; et al. Palmitoylethanolamide controls reactive gliosis and exerts neuroprotective functions in a rat model of Alzheimer’s disease. Cell Death Dis. 2014, 5, e1419.
- Scuderi, C.; Valenza, M.; Stecca, C.; Esposito, G.; Carratù, M.R.; Steardo, L. Palmitoylethanolamide exerts neuroprotective effects in mixed neuroglial cultures and organotypic hippocampal slices via peroxisome proliferator-activated receptor-α. J. Neuroinflamm. 2012, 9, 49.
- Paterniti, I.; Impellizzeri, D.; Crupi, R.; Morabito, R.; Campolo, M.; Esposito, E.; Cuzzocrea, S. Molecular evidence for the involvement of PPAR-δ and PPAR-γ in anti-inflammatory and neuroprotective activities of palmitoylethanolamide after spinal cord trauma. J. Neuroinflamm. 2013, 10, 20.
- Cristiano, C.; Pirozzi, C.; Coretti, L.; Cavaliere, G.; Lama, A.; Russo, R.; Lembo, F.; Mollica, M.P.; Meli, R.; Calignano, A.; et al. Palmitoylethanolamide counteracts autistic-like behaviours in BTBR T+tf/J mice: Contribution of central and peripheral mechanisms. Brain Behav. Immun. 2018, 74, 166–175.
- Scuderi, C.; Esposito, G.; Blasio, A.; Valenza, M.; Arietti, P.; Steardo, L., Jr.; Carnuccio, R.; De Filippis, D.; Petrosino, S.; Iuvone, T.; et al. Palmitoylethanolamide counteracts reactive astrogliosis induced by β-amyloid peptide. J. Cell Mol. Med. 2011, 15, 2664–2674.
- Scuderi, C.; Steardo, L. Neuroglial roots of neurodegenerative diseases: Therapeutic potential of palmitoylethanolamide in models of Alzheimer’s disease. CNS Neurol. Disord. Drug Targets 2013, 12, 62–69.
- D’Agostino, G.; Russo, R.; Avagliano, C.; Cristiano, C.; Meli, R.; Calignano, A. Palmitoylethanolamide protects against the amyloid-β25-35-induced learning and memory impairment in mice, an experimental model of Alzheimer disease. Neuropsychopharmacology 2012, 37, 1784–1792.
- Pertwee, R.G. GPR55: A new member of the cannabinoid receptor clan? Br. J. Pharm. 2007, 152, 984–986.
- Mattace Raso, G.; Russo, R.; Calignano, A.; Meli, R. Palmitoylethanolamide in CNS health and disease. Pharm. Res. 2014, 86, 32–41.
- Petrosino, S.; Di Marzo, V. The pharmacology of palmitoylethanolamide and first data on the therapeutic efficacy of some of its new formulations. Br. J. Pharm. 2017, 174, 1349–1365.
- De Petrocellis, L.; Davis, J.B.; Di Marzo, V. Palmitoylethanolamide enhances anandamide stimulation of human vanilloid VR1 receptors. FEBS Lett. 2001, 506, 253–256.
- Smart, D.; Jonsson, K.O.; Vandevoorde, S.; Lambert, D.M.; Fowler, C.J. ‘Entourage’ effects of N-acyl ethanolamines at human vanilloid receptors. Comparison of effects upon anandamide-induced vanilloid receptor activation and upon anandamide metabolism. Br. J. Pharm. 2002, 136, 452–458.
- Ambrosino, P.; Soldovieri, M.V.; Russo, C.; Taglialatela, M. Activation and desensitization of TRPV1 channels in sensory neurons by the PPARα agonist palmitoylethanolamide. Br. J. Pharm. 2013, 168, 1430–1444.
- Capasso, R.; Orlando, P.; Pagano, E.; Aveta, T.; Buono, L.; Borrelli, F.; Di Marzo, V.; Izzo, A.A. Palmitoylethanolamide normalizes intestinal motility in a model of post-inflammatory accelerated transit: Involvement of CB₁ receptors and TRPV1 channels. Br. J. Pharm. 2014, 171, 4026–4037.
- Snaidero, N.; Simons, M. Myelination at a glance. J. Cell Sci. 2014, 127, 2999–3004.
- Simons, M.; Nave, K.A. Oligodendrocytes: Myelination and Axonal Support. Cold Spring Harb. Perspect. Biol. 2015, 8, a020479.
- Gensert, J.M.; Goldman, J.E. Endogenous progenitors remyelinate demyelinated axons in the adult CNS. Neuron 1997, 19, 197–203.
- Fernandez-Castaneda, A.; Gaultier, A. Adult oligodendrocyte progenitor cells—Multifaceted regulators of the CNS in health and disease. Brain Behav. Immun. 2016, 57, 1–7.
- Stadelmann, C.; Timmler, S.; Barrantes-Freer, A.; Simons, M. Myelin in the Central Nervous System: Structure, Function, and Pathology. Physiol. Rev. 2019, 99, 1381–1431.
- Murtie, J.C.; Zhou, Y.X.; Le, T.Q.; Vana, A.C.; Armstrong, R.C. PDGF and FGF2 pathways regulate distinct oligodendrocyte lineage responses in experimental demyelination with spontaneous remyelination. Neurobiol. Dis. 2005, 19, 171–182.
- Kuhn, S.; Gritti, L.; Crooks, D.; Dombrowski, Y. Oligodendrocytes in Development, Myelin Generation and Beyond. Cells 2019, 8, 1424.
- Kitada, M.; Rowitch, D.H. Transcription factor co-expression patterns indicate heterogeneity of oligodendroglial subpopulations in adult spinal cord. Glia 2006, 54, 35–46.
- Hartline, D.K. What is myelin? Neuron Glia Biol. 2008, 4, 153–163.
- Fields, R.D.; Burnstock, G. Purinergic signalling in neuron-glia interactions. Nat. Rev. NeuroSci. 2006, 7, 423–436.
- Readhead, C.; Popko, B.; Takahashi, N.; Shine, H.D.; Saavedra, R.A.; Sidman, R.L.; Hood, L. Expression of a myelin basic protein gene in transgenic shiverer mice: Correction of the dysmyelinating phenotype. Cell 1987, 48, 703–712.
- Roach, A.; Takahashi, N.; Pravtcheva, D.; Ruddle, F.; Hood, L. Chromosomal mapping of mouse myelin basic protein gene and structure and transcription of the partially deleted gene in shiverer mutant mice. Cell 1985, 42, 149–155.
- Snaidero, N.; Möbius, W.; Czopka, T.; Hekking, L.H.; Mathisen, C.; Verkleij, D.; Goebbels, S.; Edgar, J.; Merkler, D.; Lyons, D.A.; et al. Myelin membrane wrapping of CNS axons by PI(3,4,5)P3-dependent polarized growth at the inner tongue. Cell 2014, 156, 277–290.
- Simons, M.; Krämer, E.M.; Thiele, C.; Stoffel, W.; Trotter, J. Assembly of myelin by association of proteolipid protein with cholesterol- and galactosylceramide-rich membrane domains. J. Cell Biol. 2000, 151, 143–154.
- Snaidero, N.; Velte, C.; Myllykoski, M.; Raasakka, A.; Ignatev, A.; Werner, H.B.; Erwig, M.S.; Möbius, W.; Kursula, P.; Nave, K.A.; et al. Antagonistic Functions of MBP and CNP Establish Cytosolic Channels in CNS Myelin. Cell Rep. 2017, 18, 314–323.
- Philips, T.; Rothstein, J.D. Oligodendroglia: Metabolic supporters of neurons. J. Clin. Investig. 2017, 127, 3271–3280.
- Traiffort, E.; Kassoussi, A.; Zahaf, A.; Laouarem, Y. Astrocytes and Microglia as Major Players of Myelin Production in Normal and Pathological Conditions. Front. Cell NeuroSci. 2020, 14, 79.
- Müller, E. Die multiple Sklerose des Gehirns und Rückenmarks: Ihre Pathologie und Behandlung. Pathol. Anat. Und. Pathog. 1904, 300–344.
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35.
- Barnett, S.C.; Linington, C. Myelination: Do astrocytes play a role? Neuroscientist 2013, 19, 442–450.
- Lundgaard, I.; Osório, M.J.; Kress, B.T.; Sanggaard, S.; Nedergaard, M. White matter astrocytes in health and disease. Neuroscience 2014, 276, 161–173.
- Verkhratsky, A.; Nedergaard, M. Physiology of Astroglia. Physiol. Rev. 2018, 98, 239–389.
- Verkhratsky, A.; Nedergaard, M.; Hertz, L. Why are astrocytes important? NeuroChem. Res. 2015, 40, 389–401.
- Verkhratsky, A.; Parpura, V.; Li, B.; Scuderi, C. Astrocytes: The Housekeepers and Guardians of the CNS. Adv. Neurobiol. 2021, 26, 21–53.
- Domingues, H.S.; Portugal, C.C.; Socodato, R.; Relvas, J.B. Oligodendrocyte, Astrocyte, and Microglia Crosstalk in Myelin Development, Damage, and Repair. Front. Cell Dev. Biol. 2016, 4, 71.
- Orthmann-Murphy, J.L.; Abrams, C.K.; Scherer, S.S. Gap junctions couple astrocytes and oligodendrocytes. J. Mol. NeuroSci. 2008, 35, 101–116.
- Nutma, E.; van Gent, D.; Amor, S.; Peferoen, L.A.N. Astrocyte and Oligodendrocyte Cross-Talk in the Central Nervous System. Cells 2020, 9, 600.
- Nagy, J.I.; Ionescu, A.V.; Lynn, B.D.; Rash, J.E. Connexin29 and connexin32 at oligodendrocyte and astrocyte gap junctions and in myelin of the mouse central nervous system. J. Comp. Neurol. 2003, 464, 356–370.
- Markoullis, K.; Sargiannidou, I.; Gardner, C.; Hadjisavvas, A.; Reynolds, R.; Kleopa, K.A. Disruption of oligodendrocyte gap junctions in experimental autoimmune encephalomyelitis. Glia 2012, 60, 1053–1066.
- Richardson, W.D.; Pringle, N.; Mosley, M.J.; Westermark, B.; Dubois-Dalcq, M. A role for platelet-derived growth factor in normal gliogenesis in the central nervous system. Cell 1988, 53, 309–319.
- McKinnon, R.D.; Waldron, S.; Kiel, M.E. PDGF alpha-receptor signal strength controls an RTK rheostat that integrates phosphoinositol 3’-kinase and phospholipase Cgamma pathways during oligodendrocyte maturation. J. NeuroSci. 2005, 25, 3499–3508.
- Moore, C.S.; Milner, R.; Nishiyama, A.; Frausto, R.F.; Serwanski, D.R.; Pagarigan, R.R.; Whitton, J.L.; Miller, R.H.; Crocker, S.J. Astrocytic tissue inhibitor of metalloproteinase-1 (TIMP-1) promotes oligodendrocyte differentiation and enhances CNS myelination. J. NeuroSci. 2011, 31, 6247–6254.
- Siegel, G.J.; Agranoff, B.W.; Albers, R.W.; Fisher, S.K.; Uhler, M.D. Basic Neurochemistry: Molecular, Cellular and Medical Aspects, 6th ed.; Lippincott-Raven: New York, NY, USA, 1999.
- Pfrieger, F.W.; Ungerer, N. Cholesterol metabolism in neurons and astrocytes. Prog. Lipid Res. 2011, 50, 357–371.
- Sofroniew, M.V. Astrocyte Reactivity: Subtypes, States, and Functions in CNS Innate Immunity. Trends Immunol. 2020, 41, 758–770.
- Bronzuoli, M.R.; Facchinetti, R.; Valenza, M.; Cassano, T.; Steardo, L.; Scuderi, C. Astrocyte Function Is Affected by Aging and Not Alzheimer’s Disease: A Preliminary Investigation in Hippocampi of 3xTg-AD Mice. Front. Pharm. 2019, 10, 644.
- Valenza, M.; Steardo, L., Jr.; Steardo, L.; Verkhratsky, A.; Scuderi, C. Systemic Inflammation and Astrocyte Reactivity in the Neuropsychiatric Sequelae of COVID-19: Focus on Autism Spectrum Disorders. Front. Cell NeuroSci. 2021, 15, 748136.
- Steardo, L., Jr.; Steardo, L.; Verkhratsky, A.; Scuderi, C. Post-COVID-19 neuropsychiatric syndrome: Is maladaptive glial recovery to blame? Acta Physiol. 2021, 233, e13717.
- Scuderi, C.; Verkhratsky, A.; Parpura, V.; Li, B. Neuroglia in Psychiatric Disorders. Adv. Neurobiol. 2021, 26, 3–19.
- Messersmith, D.J.; Murtie, J.C.; Le, T.Q.; Frost, E.E.; Armstrong, R.C. Fibroblast growth factor 2 (FGF2) and FGF receptor expression in an experimental demyelinating disease with extensive remyelination. J. NeuroSci. Res. 2000, 62, 241–256.
- Albrecht, P.J.; Dahl, J.P.; Stoltzfus, O.K.; Levenson, R.; Levison, S.W. Ciliary neurotrophic factor activates spinal cord astrocytes, stimulating their production and release of fibroblast growth factor-2, to increase motor neuron survival. Exp. Neurol. 2002, 173, 46–62.
- Selmaj, K.; Raine, C.S.; Cannella, B.; Brosnan, C.F. Identification of lymphotoxin and tumor necrosis factor in multiple sclerosis lesions. J. Clin. Investig. 1991, 87, 949–954.
- Cammer, W.; Zhang, H. Maturation of oligodendrocytes is more sensitive to TNF alpha than is survival of precursors and immature oligodendrocytes. J. Neuroimmunol. 1999, 97, 37–42.
- Ding, X.; Yan, Y.; Li, X.; Li, K.; Ciric, B.; Yang, J.; Zhang, Y.; Wu, S.; Xu, H.; Chen, W.; et al. Silencing IFN-γ binding/signaling in astrocytes versus microglia leads to opposite effects on central nervous system autoimmunity. J. Immunol. 2015, 194, 4251–4264.
- De Waard, D.M.; Bugiani, M. Astrocyte-Oligodendrocyte-Microglia Crosstalk in Astrocytopathies. Front. Cell NeuroSci. 2020, 14, 608073.
- Dooves, S.; Bugiani, M.; Postma, N.L.; Polder, E.; Land, N.; Horan, S.T.; van Deijk, A.L.; van de Kreeke, A.; Jacobs, G.; Vuong, C.; et al. Astrocytes are central in the pathomechanisms of vanishing white matter. J. Clin. Investig. 2016, 126, 1512–1524.
- Li, L.; Tian, E.; Chen, X.; Chao, J.; Klein, J.; Qu, Q.; Sun, G.; Sun, G.; Huang, Y.; Warden, C.D.; et al. GFAP Mutations in Astrocytes Impair Oligodendrocyte Progenitor Proliferation and Myelination in an hiPSC Model of Alexander Disease. Cell Stem Cell 2018, 23, 239–251.e6.
More
Information
Subjects:
Neurosciences
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.2K
Revisions:
2 times
(View History)
Update Date:
20 Sep 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No