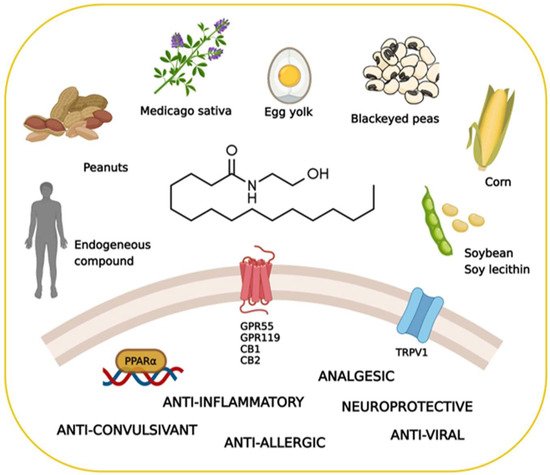
3. Myelin Sheath Organization and Functions
The term myelin was coined by Rudolf Virchow in 1864 and derives from the Greek word “myeloid” (marrow). Myelin provides the structural basis for fast impulse propagation along neuronal axons, a process required for the proper performance of motor, sensory, and cognitive functions in the CNS [
32]. The myelin sheath is an extension of the plasma membrane of mature oligodendrocytes, cells belonging to the family of glial cells [
33]. Oligodendrocytes originate from oligodendrocyte precursor cells (OPCs). During development, OPCs migrate from the ventricular/subventricular zones of the CNS to the whole brain, where they proliferate [
33]. Differentiation into oligodendrocytes was the first acknowledged function of OPCs [
34]. However, a rather large percentage of OPCs never differentiate and live throughout adulthood, suggesting that OPCs may play additional important roles [
35]. For instance, in adulthood, when a myelin lesion occurs (e.g., in cases of brain damage), local OPCs begin to proliferate and differentiate into myelinating oligodendrocytes [
34,
36].
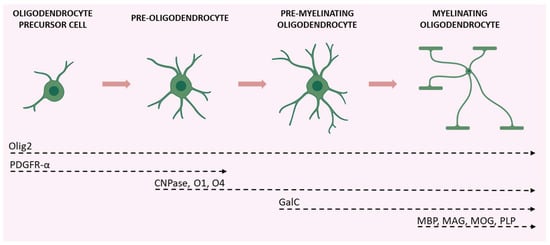
The maturation of OPCs requires several stages, each characterized by distinctive morphological and functional aspects and by the expression of specific proteins (Figure 2).
Figure 2. Schematic illustration of the various stages of OPC maturation and corresponding markers. Olig2 is expressed in all stages of the lineage; OPCs and pre-oligodendrocytes are characterized by PDGFR-α expression; CNPase, O1, and O4 are expressed during transition from pre-oligodendrocytes to differentiated oligodendrocytes (which also express GalC); axon-myelinating oligodendrocytes are characterized by myelin protein expression (MBP, MAG, MOG, and PLP). Created with
BioRender.com (accessed on 19 August 2022).
Early OPCs are bipolar cells characterized mainly by the expression of the platelet-derived growth factor receptor-α (PDGFR-α). This receptor mediates the signal of the neuronal and astrocytic PDGF-α, which regulates the proliferation, migration, survival, and maturation of OPCs [
37]. Once these cells keep contact with a target axon, they lose their bipolarity and start to develop filamentous myelin outgrowths, developing a complex shape. At this differentiation stage, pre-oligodendrocytes express 2′, 3′-cyclic-nucleotide 3′-phosphodiesterase (CNPase) and the cell surface markers O4 and O1. Finally, once fully mature, oligodendrocytes show a complex and ramified structure and form myelin sheaths. Myelinating oligodendrocytes can be identified by the expression of several markers including the myelin basic protein (MBP), the proteolipid protein (PLP), and the myelin-associated glycoprotein (MAG), as well as by the membrane marker galactocerebroside (GalC) and the surface marker myelin-oligodendrocyte glycoprotein (MOG) [
38]. Olig2 is a transcription factor that drives oligodendrocyte-lineage cells to express myelin-related genes. It appears to be constantly expressed by oligodendrocytes regardless of the degree of differentiation, but progressively decreases at maturity [
39].
The myelin structure consists of a multilayered battery of membranes organized in alternating electron-dense and electron-light layers [
40]. The electron-dense layers represent the zone of adhesion between the tightly condensed cytoplasmic membranes, while the electron-light layers consist of extracellularly arranged myelin membranes [
40]. The myelinated portions along axons are separated by the nodes of Ranvier, small myelin-free areas that promote rapid saltatory transmission of the nerve impulse by concentrating voltage-dependent sodium channels [
41]. Myelin structure is stabilized by a multitude of adhesion mechanisms and proteins. The compaction of myelin is primarily performed by MBP, which condensates two adjacent cytoplasmic membrane surfaces into a major dense line of myelin [
42,
43]. The compaction begins in the very outer layers, close to the cell soma, and proceeds to the inner side in parallel with the growth of the membrane. In order to ensure the success of this process, MBP mRNA is trafficked from the nucleus to the myelin compartment where it is locally translated [
44]. The integral membrane protein PLP allows the extracellular and cytoplasmic membranes to adhere to each other, enhancing the compaction of myelin [
45]. The presence of the cytoplasm-rich compartments within myelin is ensured by CNPase which, by directly associating with cytoskeletal actin, antagonizes the adhesive forces exerted by polymerizing MBP [
46]. Cytoplasmic channels allow the bidirectional movement of macromolecules under the myelin sheath maintaining vital such a compacted structure.
Through such channels, oligodendrocytes provide neurons with trophic and metabolic support. For example, oligodendrocytes use monocarboxylate transporters to deliver energy metabolites, like pyruvate and lactate, to neurons that use them to produce ATP [
47].
4. The Role of the Astrocyte-Oligodendrocyte Cross-Talk in Myelination
The proliferation and differentiation of OPCs need the support of other glial cells, mainly microglia and astrocytes that release distinct patterns of secreted molecules to drive these processes. Microglia involvement in OPC differentiation, as well as in the context of myelin repair, is acknowledged, and the data in the literature highlight a dual role of these cells, providing evidence for both trophic and detrimental roles of microglia on oligodendrocytes and myelin (for a comprehensive review see [
48]). Likewise, the role of astrocytes in the proliferation of OPCs and the formation and repair of myelin has long been recognized. It is believed that impairments of these star-shaped cells are implicated in the development of demyelinating diseases. This was first hypothesized by Müller in 1904, who was convinced that multiple sclerosis (MS), a demyelinating disease, was characterized by astrocytic dysfunction [
49]. Research findings collected so far have confirmed this hypothesis, with various studies highlighting the key role of the cross-talk between astrocytes and oligodendrocytes in myelination, in both health and disease [
50,
51].
Astrocytes are the most abundant glial cell type of the CNS, found in both white and grey matter [
52]. They were long considered secondary to neurons and were defined, from the Greek term glia, as “brain glue”. However, research studies over the past two decades have demonstrated that astrocytes exert a plethora of different functions to maintain CNS homeostasis at molecular, cellular, organ, and system levels of organization [
53]. Astrocytes provide both physical and metabolic support to neurons [
50] and modulate synaptic transmission and information processing by neural circuits [
54]. Astrocytes are key components of the BBB; in this way, they regulate cerebral blood flow and the communication between the CNS and the periphery [
54]. Furthermore, they are involved in several processes, such as ion and water transport, pH buffering, neuroplasticity, and synapse pruning. They also release approximately 200 molecules, including neurotrophic factors and energy substrates, thus, providing trophic and metabolic support to all cells in the CNS [
55].
Astrocytes and oligodendrocytes originate from a common lineage of neural progenitor cells within the neuroectoderm [
56] and, after development, communicate in physical and functional ways. Physically, astrocytes are coupled to oligodendrocytes through gap junctions at the cell body level and the paranodes, directly connecting the outer layer of the myelin sheath with an astrocytic process [
57,
58]. Such intercellular channels are heterologous, since oligodendrocytes express connexin (Cx) 32, Cx47, and Cx29, whereas astrocytes have Cx26, Cx30, and Cx43 [
59]. A large number of reports underscore the importance of gap junctions in myelination. For instance, mutations or genetic ablation of specific connexins could lead to myelin defects, demyelinating diseases, encephalopathies, and peripheral neuropathies [
60].
Besides direct physical interaction, astrocytes can communicate with oligodendrocytes by releasing a variety of soluble factors, including PDGF, brain-derived neurotrophic factor (BDNF), ciliary neurotrophic factor (CNTF), transforming growth factor (TGF)-β, and basic fibroblast growth factor (FGF2). All of these are considered promoters of OPC proliferation and survival [
48], despite some contrasting evidence suggesting that PDGF and FGF2 could also act as inhibitors [
61,
62]. Moreover, the tissue inhibitor of metalloproteinase-1 (TIMP-1), an endogenous regulator of matrix metalloproteinases, promotes oligodendrogenesis [
63]. In addition, astrocytes maintain and support myelin sheath formation by synthesizing and delivering cholesterol to oligodendrocytes [
64,
65].
As astrocytes are critical for maintaining brain homeostasis [
50], they promptly react to any CNS insult or damage by undergoing morphofunctional changes that are context-, time-, and disease-specific [
66,
67,
68,
69]. Astrocyte reactivity could have either beneficial or detrimental consequences for myelination, and the difference seems to be related to the severity of astrogliosis [
70]. Mild astrogliosis leads to the release of CNTF, FGF2, and the proinflammatory interleukin (IL)-6, all associated with OPC survival, proliferation, and maturation [
71,
72]. During severe astrogliosis, astrocytes secrete tumor necrosis factor (TNF)-α, which correlates with the extent of demyelination in MS and with myelin and oligodendrocytes damage in vitro [
73,
74]. Additionally, in an animal model of experimental autoimmune encephalomyelitis (EAE) the release of interferon (IFN)-γ was shown to suppress remyelination and delay recovery [
75].
The relevance of astrocyte contribution to oligodendrocyte function is also evident in several astrocytopathies, such as Alexander disease, vanishing white matter, megalencephalic leukoencephalopathy with subcortical cysts, Aicardi–Goutières syndrome, and oculodentodigital dysplasia [
76]. All these diseases are characterized by genetic mutations that cause defective astrocyte function, with major consequences for oligodendrocyte physiology as low oligodendrocyte survival, impaired myelination, and absence of myelin with or without concurrent development of astrogliosis [
77,
78].
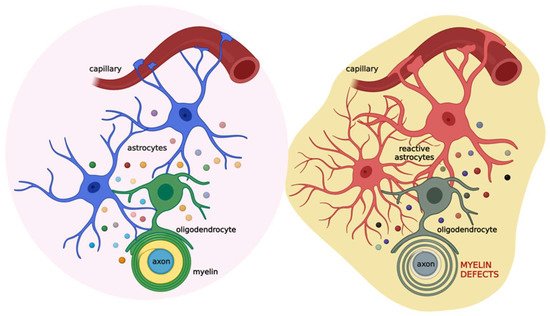
Based on this evidence, astrocytes appear as the major cells orchestrating cell-to-cell communication relevant for myelination. Thus, alterations in this cross-talk could lead to myelin defects (Figure 3).
Figure 3. Schematic illustration of the coupling of oligodendrocytes, astrocytes, and neurons in physiological (left) and pathological (right) conditions. Astrocytes (blue) communicate with oligodendrocytes (green) through gap junctions and several released factors (colored circles), such as PDGF, BDNF, CNTF, TGF-β, FGF2, TIMP-1, ILs, TNF-α, and IFN-γ. This cross-talk allows the proper maturation of oligodendrocytes and their ability to form the myelin sheath. This, in turn, impacts neuronal activity. When brain damage occurs (see text for details), astrocytes (red) react promptly, affecting oligodendrocytes (grey) activity and ultimately the myelin sheath. Created with
BioRender.com (accessed on 22 July 2022).