 +1 credit
+1 credit
Video Upload Options
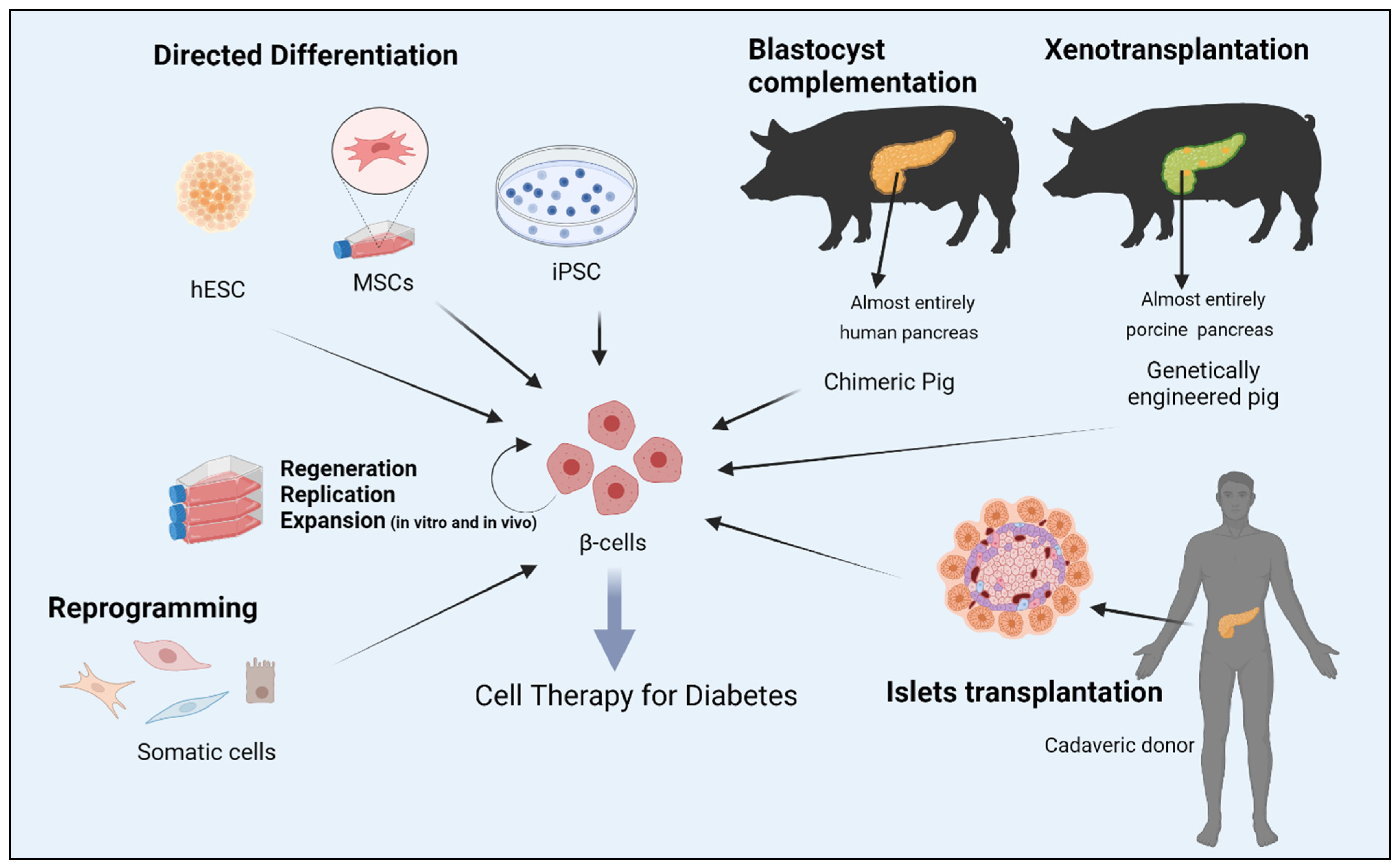
Numerous cell sources are being explored to replenish functional β-cell mass since the proof-of -concept for cell therapy of diabetes was laid down by transplantation of islets. Various strategies that aim to generate bone fide insulin producing cells are explored. In particular on reprogramming and especially on α-cells conversion into insulin producing cells are focused here. A logical place to begin with for generating β-cells is to utilise the plasticity of closely related endoderm derived cell types like pancreatic non-β-cells and coaxing them to adopt a β-cell phenotype. Given the close ontogenetic relationship, functional similarity and dependency among these cells, the potential for interconversion is unequivocal. Phenotypic plasticity between pancreatic α-cells and β-cells is notably pronounced.
1. Introduction
2. Reprogramming Cells to Make Insulin
3. Alpha to β-cell Reprogramming
4. Conclusions and Future Perspectives
References
- International Diabetes Federation. Diabetes Facts & Figures; International Diabetes Federation: Brussels, Belgium, 2021; Available online: https://idf.org/aboutdiabetes/what-is-diabetes/facts-figures.html (accessed on 1 September 2022).
- Lin, X.; Xu, Y.; Pan, X.; Xu, J.; Ding, Y.; Sun, X.; Song, X.; Ren, Y.; Shan, P.F. Global, regional, and national burden and trend of diabetes in 195 countries and territories: An analysis from 1990 to 2025. Sci. Rep. 2020, 10, 14790.
- Chen, H.; Chen, G.; Zheng, X.; Guo, Y. Contribution of specific diseases and injuries to changes in health adjusted life expectancy in 187 countries from 1990 to 2013: Retrospective observational study. BMJ 2019, 364, l969.
- Holman, R.R.; Paul, S.K.; Bethel, M.A.; Matthews, D.R.; Neil, H.A. 10-year follow-up of intensive glucose control in type 2 diabetes. N. Engl. J. Med. 2008, 359, 1577–1589.
- Diabetes Control and Complications Trial (DCCT); Epidemiology of Diabetes Interventions and Complications (EDIC) Research Group. Effect of intensive diabetes therapy on the progression of diabetic retinopathy in patients with type 1 diabetes: 18 years of follow-up in the DCCT/EDIC. Diabetes 2015, 64, 631–642.
- Zinman, B.; Genuth, S.; Nathan, D.M. The Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Study: 30th Anniversary Presentations. Diabetes Care 2014, 37, 8.
- Polonsky, K.S. The Past 200 Years in Diabetes. N. Engl. J. Med. 2012, 367, 1332–1340.
- NICE-Sugar Study Investigators. Hypoglycemia and risk of death in critically ill patients. N. Engl. J. Med. 2012, 367, 1108–1118.
- Bluestone, J.A.; Herold, K.; Eisenbarth, G. Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature 2010, 464, 1293–1300.
- Brown, S.A.; Kovatchev, B.P.; Raghinaru, D.; Lum, J.W.; Buckingham, B.A.; Kudva, Y.C.; Laffel, L.M.; Levy, C.J.; Pinsker, J.E.; Wadwa, R.P.; et al. Six-Month Randomized, Multicenter Trial of Closed-Loop Control in Type 1 Diabetes. N. Engl. J. Med. 2019, 381, 1707–1717.
- Shapiro, A.M.J.; Ricordi, C.; Hering, B.J.; Auchincloss, H.; Lindblad, R.; Robertson, R.P.; Secchi, A.; Brendel, M.D.; Berney, T.; Brennan, D.C.; et al. International Trial of the Edmonton Protocol for Islet Transplantation. N. Engl. J. Med. 2006, 355, 1318–1330.
- Oliver-Krasinski, J.M.; Stoffers, D.A. On the origin of the beta cell. Genes Dev. 2008, 22, 1998–2021.
- Assady, S.; Maor, G.; Amit, M.; Itskovitz-Eldor, J.; Skorecki, K.L.; Tzukerman, M. Insulin Production by Human Embryonic Stem Cells. Diabetes 2001, 50, 1691–1697.
- D’Amour, K.A.; Agulnick, A.D.; Eliazer, S.; Kelly, O.G.; Kroon, E.; Baetge, E.E. Efficient differentiation of human embryonic stem cells to definitive endoderm. Nat. Biotechnol. 2005, 23, 1534–1541.
- D’Amour, K.A.; Bang, A.G.; Eliazer, S.; Kelly, O.G.; Agulnick, A.D.; Smart, N.G.; Moorman, M.A.; Kroon, E.; Carpenter, M.K.; Baetge, E.E. Production of pancreatic hormone–expressing endocrine cells from human embryonic stem cells. Nat. Biotechnol. 2006, 24, 1392–1401.
- Kroon, E.; Martinson, L.A.; Kadoya, K.; Bang, A.G.; Kelly, O.G.; Eliazer, S.; Young, H.; Richardson, M.; Smart, N.G.; Cunningham, J.; et al. Pancreatic endoderm derived from human embryonic stem cells generates glucose-responsive insulin-secreting cells in vivo. Nat. Biotechnol. 2008, 26, 443–452.
- Nair, G.G.; Liu, J.S.; Russ, H.A.; Tran, S.; Saxton, M.S.; Chen, R.; Juang, C.; Li, M.-I.; Nguyen, V.Q.; Giacometti, S.; et al. Recapitulating endocrine cell clustering in culture promotes maturation of human stem-cell-derived beta cells. Nat. Cell Biol. 2019, 21, 263–274.
- Hogrebe, N.J.; Augsornworawat, P.; Maxwell, K.G.; Velazco-Cruz, L.; Millman, J.R. Targeting the cytoskeleton to direct pancreatic differentiation of human pluripotent stem cells. Nat. Biotechnol. 2020, 38, 460–470.
- Rezania, A.; Bruin, J.E.; Arora, P.; Rubin, A.; Batushansky, I.; Asadi, A.; O’Dwyer, S.; Quiskamp, N.; Mojibian, M.; Albrecht, T.; et al. Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat. Biotechnol. 2014, 32, 1121–1133.
- Pagliuca, F.W.; Millman, J.R.; Gürtler, M.; Segel, M.; Van Dervort, A.; Ryu, J.H.; Peterson, Q.P.; Greiner, D.; Melton, D.A. Generation of functional human pancreatic beta cells in vitro. Cell 2014, 159, 428–439.
- Russ, H.A.; Parent, A.V.; Ringler, J.J.; Hennings, T.G.; Nair, G.G.; Shveygert, M.; Guo, T.; Puri, S.; Haataja, L.; Cirulli, V.; et al. Controlled induction of human pancreatic progenitors produces functional beta-like cells in vitro. EMBO J. 2015, 34, 1759–1772.
- Naujok, O.; Burns, C.; Jones, P.M.; Lenzen, S. Insulin-producing surrogate beta-cells from embryonic stem cells: Are we there yet? Mol. Ther. 2011, 19, 1759–1768.
- Pagliuca, F.W.; Melton, D.A. How to make a functional beta-cell. Development 2013, 140, 2472–2483.
- Hamaguchi, K.; Utsunomiya, N.; Takaki, R.; Yoshimatsu, H.; Sakata, T. Cellular interaction between mouse pancreatic alpha-cell and beta-cell lines: Possible contact-dependent inhibition of insulin secretion. Exp. Biol. Med. 2003, 228, 1227–1233.
- Thorel, F.; Népote, V.; Avril, I.; Kohno, K.; Desgraz, R.; Chera, S.; Herrera, P.L. Conversion of adult pancreatic alpha-cells to beta-cells after extreme beta-cell loss. Nature 2010, 464, 1149–1154.
- Collombat, P.; Xu, X.; Ravassard, P.; Sosa-Pineda, B.; Dussaud, S.; Billestrup, N.; Madsen, O.D.; Serup, P.; Heimberg, H.; Mansouri, A. The ectopic expression of Pax4 in the mouse pancreas converts progenitor cells into alpha and subsequently beta cells. Cell 2009, 138, 449–462.
- Yang, Y.P.; Thorel, F.; Boyer, D.F.; Herrera, P.L.; Wright, C.V. Context-specific alpha- to-beta-cell reprogramming by forced Pdx1 expression. Genes Dev. 2011, 25, 1680–1685.
- Matsuoka, T.A.; Kawashima, S.; Miyatsuka, T.; Sasaki, S.; Shimo, N.; Katakami, N.; Kawamori, D.; Takebe, S.; Herrera, P.L.; Kaneto, H.; et al. Mafa Enables Pdx1 to Effectively Convert Pancreatic Islet Progenitors and Committed Islet alpha-Cells Into beta-Cells In Vivo. Diabetes 2017, 66, 1293–1300.
- Chakravarthy, H.; Gu, X.; Enge, M.; Dai, X.; Wang, Y.; Damond, N.; Downie, C.; Liu, K.; Wang, J.; Xing, Y.; et al. Converting Adult Pancreatic Islet alpha Cells into beta Cells by Targeting Both Dnmt1 and Arx. Cell Metab. 2017, 25, 622–634.
- Furuyama, K.; Chera, S.; Van Gurp, L.; Oropeza, D.; Ghila, L.; Damond, N.; Vethe, H.; Paulo, J.A.; Joosten, A.M.; Berney, T.; et al. Diabetes relief in mice by glucose-sensing insulin-secreting human alpha-cells. Nature 2019, 567, 43–48.
- Xiao, X.; Guo, P.; Shiota, C.; Zhang, T.; Coudriet, G.M.; Fischbach, S.; Prasadan, K.; Fusco, J.; Ramachandran, S.; Witkowski, P.; et al. Endogenous Reprogramming of Alpha Cells into Beta Cells, Induced by Viral Gene Therapy, Reverses Autoimmune Diabetes. Cell Stem Cell 2018, 22, 78–90.e4.
- Fomina-Yadlin, D.; Kubicek, S.; Walpita, D.; Dančik, V.; Hecksher-Sørensen, J.; Bittker, J.A.; Sharifnia, T.; Shamji, A.; Clemons, P.A.; Wagner, B.K.; et al. Small-molecule inducers of insulin expression in pancreatic α-cells. Proc. Natl. Acad. Sci. USA 2010, 107, 15099–15104.
- Lee, Y.S.; Lee, C.; Choung, J.S.; Jung, H.S.; Jun, H.S. Glucagon-Like Peptide 1 Increases beta-Cell Regeneration by Promoting alpha- to beta-Cell Transdifferentiation. Diabetes 2018, 67, 2601–2614.
- Ben-Othman, N.; Vieira, A.; Courtney, M.; Record, F.; Gjernes, E.; Avolio, F.; Hadzic, B.; Druelle, N.; Napolitano, T.; Navarro-Sanz, S.; et al. Long-Term GABA Administration Induces Alpha Cell-Mediated Beta-like Cell Neogenesis. Cell 2017, 168, 73–85.e11.
- Menegaz, D.; Hagan, D.W.; Almaça, J.; Cianciaruso, C.; Rodriguez-Diaz, R.; Molina, J.; Dolan, R.M.; Becker, M.W.; Schwalie, P.C.; Nano, R.; et al. Mechanism and effects of pulsatile GABA secretion from cytosolic pools in the human beta cell. Nat. Metab. 2019, 1, 1110–1126.
- Li, J.; Casteels, T.; Frogne, T.; Ingvorsen, C.; Honoré, C.; Courtney, M.; Huber, K.V.; Schmitner, N.; Kimmel, R.A.; Romanov, R.A.; et al. Artemisinins Target GABAA Receptor Signaling and Impair α Cell Identity. Cell 2016, 168, 86–100.e15.
- van der Meulen, T.L.S.; Noordeloos, E.; Donaldson, C.J.; Adams, M.W.; Noguchi, G.M.; Mawla, A.M.; Huising, M.O. Artemether Does Not Turn α Cells into β Cells. Cell Metab. 2018, 27, 218–225.e4.
- Ackermann, A.M.; Moss, N.G.; Kaestner, K.H. GABA and Artesunate Do Not Induce Pancreatic alpha-to-beta Cell Transdifferentiation In Vivo. Cell Metab. 2018, 28, 787–792.e3.
- Wang, M.Y.; Dean, E.D.; Quittner-Strom, E.; Zhu, Y.; Chowdhury, K.H.; Zhang, Z.; Zhao, S.; Li, N.; Ye, R.; Lee, Y.; et al. Glucagon blockade restores functional beta-cell mass in type 1 diabetic mice and enhances function of human islets. Proc. Natl. Acad. Sci. USA 2021, 118, e2022142118.


